

1 引言
孤独症谱系障碍(Autism Spectrum Disorders, ASD), 简称孤独症, 是一种发病于婴幼儿时期的、常见的社会性发展障碍, 与大脑的神经化学机制异常有着密切的关系。美国精神疾病手册第五版(Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, DSM-V)指出孤独症患者的核心症状表现为:持续性的社会交流和社会互动能力缺失, 以及兴趣狭窄和重复刻板的行为方式。美国疾病控制与预防中心(Christensen et al., 2012)最新调查结果显示, 儿童孤独症患病率已达14.4‰, 即每68名8岁以下儿童中就有一名孤独症患儿, 与2000年相比, 患病比率增长了2.18倍。因此, 探究孤独症的发病原因已经成为医学、生物学界的重要议题之一。
生物遗传学研究表明, 大约10%~30%孤独症发病是由基因异常导致的(Huguet, Ey, & Bourgeron, 2013; Gaugler et al., 2014; Sanders et al., 2015), 即基因异常影响了其编码的蛋白质的结构和功能, 进而改变了脑的特定功能, 最终表现为患者的认知和行为异常。双生子研究也证明遗传因素在孤独症发病中起着非常重要的作用, 同卵双生的孤独症共患率大约为77%~95%, 显著高于异卵双生子31% (Ronald, Happe, & Plomin, 2005; Taniai, Nishiyama, Miyachi, Imaeda, & Sumi, 2008; Rosenberg et al., 2009)。家族聚集性研究显示, 同胞患孤独症的几率为10%~20%, 大约是家庭中出现新生孤独症概率的20倍(Ozonoff et al., 2011; Wood et al., 2015), 据此推测父母某一方患孤独症其子代患病风险大概为10%~15%, 且男婴患病率高于女婴(Vorstman et al., 2017)。根据同卵双生、异卵双生共患的差异以及患者同胞再患的危险度推断, 孤独症的遗传几率可达91%~93% (Bailey et al., 1995)。借助基因二代测序技术, 已发现多个染色体区域上的拷贝数变异(Copy Number Variants, CNV)会增加孤独症患病风险。到目前为止, 有4%~20% 的孤独症患者携带疾病相关的CNV (Schaaf & Zoghbi, 2011; Pinto et al., 2014), 已发现的包含CNV的染色体片段达2223个, 遍及所有染色体。除此之外, 基因新生突变(de novo mutations)也被认为是孤独症发生的一个重要原因。SFARI (Simons Foundation Autism Research Initiative)目前已经收录了990个孤独症相关基因, 包括SHANK3, SYNGAP1, NRXN1, NLGN3/4X, CNTN4, NLGN1, UBE3A, SCN2A, RELN和CHD8等(Michaelson et al., 2012; Pinto et al., 2011; Roohi et al., 2008; Neale et al., 2012; Bernier et al., 2014)。其中部分已经实验验证为孤独症易感基因, 如SHANK3基因突变影响神经元突触发育过程, 导致该基因缺失小鼠表现出多项典型的孤独症行为特征(Durand et al., 2007)。CHD8无义突变使转录过程提前终止, 导致编码产物缩短, 破坏了蛋白质原有功能, 影响神经元增殖、树突发育和突触形成过程, 被认为是导致孤独症发病的重要风险因素(Bernier et al., 2014; O’Roak, Vives, Fu, et al., 2012; Neale et al., 2012)。AUTS2基因突变改变了对组蛋白H2A的化学修饰, 使得小鼠出现孤独症类似行为(Gao et al., 2014)。此类研究均证实了基因功能异常是孤独症发生的重要原因。
目前已发现的孤独症易感基因多与神经系统发育有关, 涉及神经细胞的运动与增殖、神经元的轴突投射、树突棘可塑性、突触形成和维持等, 与核染色质重组、基因转录调控、酶的活性调控、细胞骨架调控、蛋白化学修饰等过程密切相关(Pinto et al., 2010; Sanders et al., 2012; Sakai et al., 2011; O’Roak, Vives, Fu, et al., 2012; King et al., 2013; Donato, Chowdhury, Lahr, & Caroni, 2015), 所涉及的分子信号通路包括Wnt信号通路(O’Roak, Vives, Girirajan, et al., 2012; Mine, Yuskaitis, King, Beurel, & Jope, 2010; Okerlund & Cheyette, 2011)、钙离子信号通路(Yun & Trommer, 2011; Moretti et al., 2006)、神经生长因子(nerve growth factor, NGF)信号通路(Riikonen & Vanhala, 1999; Nelson et al., 2001)、以及G蛋白偶联受体(G Protein-Coupled Receptor, GPCR)信号通路等(Zhang & Alger, 2010; Maccarrone et al., 2010; Chen et al., 2011; Silverman et al., 2012)。由此可见, 基因异常影响了关键的神经细胞信号转导, 因此被视作孤独症发生的高风险因素之一。近年来以基因为靶点开展孤独症研究已成为了相关领域研究者关注的重点。
长期以来, 人们对孤独症的认识多是从异常行为入手。有学者指出, 孤独症患者个体之间存在巨大差异, 且不同基因突变可导致不同孤独症行为特征(Happe, Ronald, & Plomin, 2006), 一些针对刻板行为和交流障碍的研究已证实了该现象(Cuccaro et al., 2003; Buxbaum et al., 2001)。所以将基因功能和行为研究联系起来, 不但能揭示孤独症发病机制, 更能促进孤独症治疗和康复(State & Sestan, 2012)。
2 毒蕈碱型乙酰胆碱受体Ⅲ亚型(cholinergic receptor, muscarinic 3, CHRM3)
作为一种神经递质, 乙酰胆碱(acetylcholine, ACh)在信号传递中扮演着重要角色, 可调节神经系统发育和神经元兴奋性变化。胆碱能神经元广泛分布于全脑, 涉及学习记忆、认知调节、情绪控制以及社会交往等过程(Bentley, Vuilleumier, Thiel, Driver, & Dolan, 2003; Dani & Bertrand, 2007; Karva & Kimchi, 2014), 胆碱能信号通路异常与多种精神类疾病的发生有关(Bowen, Smith, White, & Davison, 1976; Whitehouse et al., 1982; Deng, & Reiner., 2016)。动物模型研究发现胆碱能相关基因突变会导致小鼠出现孤独症症状(Zhang et al., 2016), 基因功能异常影响脑内胆碱能信号通路的信号传递以及胆碱能相关因子的表达水平, 进而引发孤独症。同时, 还有研究发现孤独症患者脑内灰质和颞叶脑区胆碱能信号通路异常(Perry et al., 2001; Lee et al., 2002; Martin-Ruiz et al., 2004; Ray et al., 2005; Friedman et al., 2006; Deutsch, Urbano, Neumann, Burket, & Katz, 2010; Petersen et al., 2013), 药物学研究中利用VPA (valproic acid)大鼠模型发现, 给孕期大鼠注射VPA能够导致大鼠及其子代的胆碱能神经系统紊乱, 增加患孤独症的风险, 而使用ACh酯酶抑制剂药物对缓解其出现的社交障碍、认知障碍和重复刻板行为问题十分有效(Kim er al., 2014)。目前美国食品药物管理局(Food and Drug Administration, FDA)已批准使用ACh酯酶抑制剂缓解孤独症症状(Dineley, Pandya, & Yakel, 2015), 因此, 胆碱能相关通路应在孤独症研究和治疗中受到更多关注, 检测其正常与否在未来也许可以成为研究、诊断和治疗孤独症或是区分孤独症不同亚型的一个重要参考指标。
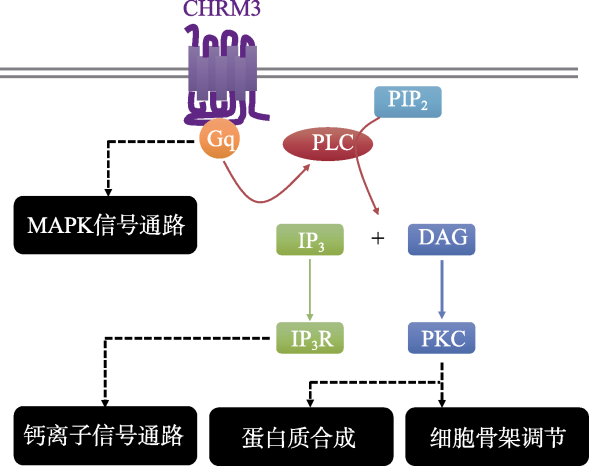
毒蕈碱型乙酰胆碱受体Ⅲ亚型(cholinergic receptor, muscarinic 3, CHRM3)是介导ACh信号传递的受体之一, 是毒蕈碱型乙酰胆碱受体(muscarinic acetylcholine receptor, mAChR)家族一员, 广泛分布于前脑、海马以及下丘脑等区域, 在脑内神经信号传导和行为调节中具有重要作用(Levey, Edmunds, Heilman, Desmond, & Frey, 1994)。CHRM3属于G蛋白偶联受体, 是一种大量分布在神经系统中的突触后膜促代谢型受体。在正常生理状况下, CHRM3接收到乙酰胆碱信号刺激后通过Gq蛋白激活磷脂酶C (PLC, phospholipase C), 进而作用于第二信使二酰甘油(DAG, diacylglycerol)和三磷酸肌醇(IP3, inositol 1, 4, 5-triphosphate), 调控细胞的增殖、代谢、细胞骨架和突触可塑性(Matsui et al., 1999)。由于CHRM3分布广泛, 对个体高级神经活动的发生有着关键性的作用, 因此CHRM3基因突变会对神经系统生长发育产生重要的影响, 可能导致癫痫(Koeleman, 2018)、精神分裂症(Devor et al., 2017)、阿尔茨海默症(Tsang et al., 2008)等多种神经系统疾病。近年来, 越来越多的研究者开始关注GPCRs以及Gq-PLC信号通路异常与孤独症的关系(Chen et al., 2011; Silverman et al., 2012; O'Connor, Bariselli, & Bellone, 2014)。遗传学研究证实, 位于Gq-PLC信号通路下游的PTEN基因是孤独症易感基因(Spinelli, Black, Berg, Eickholt, & Leslie, 2015; Cupolillo et al., 2015)。药物研究发现给孤独症模型小鼠BTBR T~(+)tf/J注射mGlu5R拮抗剂对于改善小鼠的刻板行为和社交行为有明显的效果(Silverman et al., 2012)。值得注意的是, mGlu5R与CHRM3同为G蛋白偶联受体, 均通过与Gq蛋白偶联激活PLC。这一系列研究暗示CHRM3及Gq-PLC信号通路可能对孤独症发生发展有重要影响。
临床报道与基因检测结果均表明CHRM3基因所在的1q43染色体区域缺陷与孤独症相关(见表1, Perrone et al., 2012; Petersen et al., 2013; Soueid et al., 2016)。该基因突变患者会表现出不同程度的行为异常、认知障碍、言语障碍以及运动发育迟缓等问题(Silipigni et al., 2017; Luukkonen et al., 2017)。Gai等人在(2012)年通过单核苷酸多态性微阵列(SNP microarray)技术对1224名孤独症患者的染色体进行分析, 结果显示有患者的CHRM3编码区内存在CNV (Gai et al., 2012)。此外, 利用全基因组关联分析等方法, 多项研究都提出CHRM3基因可能是孤独症易感基因(Hussman et al., 2011; De Rubeis et al., 2014; Butler, Rafi, & Manzardo, 2015; Ch'ng, Kwok, Rogic, & Pavlidis, 2015; Li et al., 2017), 从统计学角度证实了CHRM3基因突变会提高孤独症患病风险。同时研究者在动物模型研究中也发现, 抑制或过度激活CHRM3都将会导致小鼠出现不同程度的孤独症样异常行为(Alexander et al., 2009; Wang & McGinty, 1997; Amodeo, Sweeney, & Ragozzino, 2014)。上述结果说明CHRM3基因与孤独症发生之间存在密切联系。
表1 孤独症家系研究中的CHRM3突变
| 突变类型 | 等位基因改变 | 氨基酸改变 | 遗传模式 | 参考文献 |
|---|---|---|---|---|
| 无义突变 | c.1762C>T | p.Gln588Ter | 家系遗传 | Li et al., (2017) |
| 错义突变 | c.1504A>G | p.Ile502Val | 新生突变 | De Rubeis et al., (2014) |
| 错义突变 | c.1423A>T | p.Ile475Phe | 新生突变 | Li et al. (2017) |
| 缺失 | — | — | 新生突变 | Perrone et al., (2012) |
| 缺失 | — | — | 未知 | Petersen AK et al., (2012) |
图1
3 CHRM3基因异常的孤独症患者临床研究
近期已有两例与CHRM3基因异常密切相关的典型孤独症病例被相继报道。
患者一:Perrone等人(2012)报道了一名7岁的意大利男性孤独症患者。该患者为非近亲生独子, 足月分娩出生。出生体重3.4 kg, 身高34 cm, 哺乳时吸入困难, 同时伴有运动功能发育迟缓(12月龄独坐, 4岁独走)、智力低下、隐睾、身体矮小, 生长发育迟缓以及孤独症行为等特征。查体显示枕骨周围有脱发斑点, 出现脱发迹象; 脚趾拇指和第五指先天性趾侧弯; 有内斜视和咬手的问题特征; 在喂养方面由于患者有咀嚼困难的问题, 因此只能吃混合食物。基因检测结果显示患者1号染色体丢失91172 bp, 为新生突变, 该缺失区域包含RPS7P5基因、FMN2基因、CHRM3基因。其中, RPS7P5为假基因, 即在基因组上的非功能性基因组DNA拷贝, 一般情况下不被转录, 没有明确的生理意义。FMN2基因和CHRM3基因均与中枢神经系统发育有关, 是潜在的致病基因。患者MRI (Magnetic Resonance Imaging)、心电图和腹部超声检查正常。
患者二:Petersen等人(2013)报道的是一名3岁7个月的男性患者, 患者系G3P1A1 (怀孕3次; 分娩1次; 流产1次)母亲足月生胎儿, 出生体重3.3 kg。4个月时常规查体和MRI检查发现患者表现出斜视和颅神经麻痹的症状, 12个月左右被发现语言发育迟缓, 3岁7个月时经ADOS (Autism Diagnostic Observation Schedule)诊断为孤独症。患者表现出多动、易怒、注意力差、自伤行为倾向、对触觉/视觉刺激异常敏感、行为刻板、社交能力严重受损等行为缺陷。基因检测结果显示患者1号染色体丢失473 kb, 为新生突变, 丢失区域内只含有CHRM3基因。此外, 患者母亲报告在产前曾出现子痫前期的症状。
将两名CHRM3基因缺失的孤独症患者的临床表现进行对比, 发现患者均表现出认知功能受损、发育迟缓、进食困难的特征(表2)。此外, 在目前报道的其他CHRM3基因缺失的临床案例中, 患者还出现了癫痫、中风、发育迟缓以及注意力缺陷等与神经系统功能异常有关的特征(Shimojima et al., 2012; Luukkonen et al., 2017)。
表2 两名CHRM3基因缺失的孤独症患者的临床表现对比
| 特征 | Perrone等人报道的患者 | Andrea Klunder Petersen等人报道的患者 |
|---|---|---|
| 年龄、性别 | 7岁, 男 | 3岁7个月, 男 |
| 智力缺陷 | + | + |
| 发育迟缓 | + | + |
| 孤独症行为 | + | + |
| 癫痫 | - | - |
| 进食困难 | + | + |
| 身材短小 | + | - |
| 体重偏轻 | + | - |
| 曲指 | + | - |
| 斜视 | + | + |
| 自伤倾向 | + | + |
| 脑部核磁共振造影 | 正常 | 正常 |
| 社交退缩 | + | + |
| 言语发育迟缓 | + | + |
| 运动发育迟缓 | + | NA |
注:NA, date not available
4 CHRM3相关动物模型研究
4.1 CHRM3异常与孤独症刻板行为
重复刻板行为是孤独症诊断中的一项重要标准。在《精神疾病诊断与统计手册第五版》(DSM-V)中, 刻板行为被定义为:一种重复性、限制性的行为、兴趣或活动。其主要表现为自我刺激行为, 如尖叫、转圈等和自伤行为, 还包括一些仪式性、规则性的行为, 具体表现为每天在固定的时间完成某项任务, 或者固定地以某种方式进行某项活动等。刻板行为会严重影响患者的正常生活, 对患者的社交和学习造成阻碍。
Petersen等人(2013)报道的CHRM3基因异常的患者表现出刻板行为:经常抓自己的头发、用头撞墙, 只吃固定的食物; 同时患者也出现咬手的自伤行为。因此孤独症患者的刻板行为可能与CHRM3基因异常有关。在孤独症的动物模型研究中, 改变CHRM3基因的功能不仅会影响孤独症小鼠的刻板行为, 还会影响正常小鼠是否会出现孤独症样行为特征。
BTBR T~(+)tf/J (简称BTBR)小鼠是一种近交系小鼠, 即不同个体间98%以上的基因座为纯和状态的小鼠品系, 因此具有稳定的基因型。该品系小鼠能在不同代子代中稳定地表现出社会交往交流障碍和重复刻板的行为、兴趣等孤独症样行为, 以及与孤独症患者类似的脑发育异常、免疫生化指标异常的问题特征(Yang et al., 2007; Bolivar, Walters, & Phoenix, 2007), 是一种良好的孤独症研究动物模型。研究发现BTBR小鼠脑内乙酰胆碱水平显著低于野生型小鼠(McTighe, Neal, Lin, Hughes, & Smith, 2013), 给小鼠注射M型受体激动剂氧化震颤素(Oxotremorine)可以显著减少小鼠的自我理毛和埋珠子等刻板行为(Amodeo et al., 2014)。另外在临床药理学研究中也曾发现, 当给孤独症患者使用拮抗M型乙酰胆碱受体的精神类药物后, 患者重复刻板问题行为显著增加(Martin, Koenig, Scahill, & Bregman, 1999; Hardan, Jou, & Handen, 2005)。但是以上有关研究只是发现改变M型受体的信号转导功能会影响孤独症的重复刻板行为出现, 并没有详细探究这种异常是否是由于CHRM3功能异常所致。
Alexander等人(2009)的研究证明, 改变CHRM3功能将会影响小鼠出现重复刻板的孤独症样行为。研究者使改造后的人CHRM3 (human M3 muscarinic DREADD receptor coupled to Gq, hM3Dq)基因在小鼠前脑中正常表达, 由于hM3Dq无法接受内源性乙酰胆碱的信号刺激, 因此注射叠氮平-N-氧化物(clozapine-N-oxide, CNO)可以诱导激活CHRM3下游信号通路, 起到过度激活CHRM3的效果。研究者发现当不给hM3Dq小鼠注射外源性配体CNO时, hM3Dq小鼠与野生型小鼠的各项行为指标均无显著差异。当给小鼠注射较高浓度CNO后, CHRM3被过度激活, hM3Dq小鼠的刻板行为显著增加, 多动行为增多且出现癫痫症状。上述研究不仅揭示了CHRM3功能与孤独症刻板行为间的关系, 也为孤独症患者的行为干预提出了新的思路和方法。
4.2 CHRM3异常与认知功能受损
认知功能受损并非孤独症诊断标准中的核心症状, 但是绝大多数孤独症患者都伴有不同程度的认知功能受损问题(Wing, 1981; Crane, Pring, Jukes, & Goddard, 2012)。美国疾病控制与预防中心(CDCP 2012)的调查结果显示42%~60%的孤独症患者表现出认知功能受损的特征, 具体体现为患者在基本概念认知、记忆力、注意力等方面的表现低于正常儿童。缺乏正常的认知能力导致孤独症儿童无法对图形符号或语言指令做出正确的识别、理解和应答, 且由于孤独症患者均存在不同程度的语言沟通困难, 进而也无法与老师或家长进行沟通, 患者的学习过程受到了极大的阻碍。因此提高孤独症患者的认知能力有利于提高患者的生活技能、适应人际交往活动。脑发育过程中CHRM3在大脑皮层和海马等区域大量表达(Levey, Edmunds, Koliatsos, Wiley, & Heilman, 1995), 意味着CHRM3基因可能与认知功能有关。Perrone和Petersen等人报道的两例CHRM3基因变异的孤独症患者也都出现了智力发育落后、注意力缺陷等认知功能受损的问题。
Poulin等人(2010)在研究中发现, CHRM3基因敲除小鼠在恐惧性条件反射(fear conditioning)实验中依赖海马的环境联系性记忆能力均显著低于野生型小鼠。由于小鼠的痛觉和焦虑反应与野生型小鼠没有显著差异, 因此研究者推测小鼠表现出来的这种认知功能受损可能源于海马CHRM3功能异常。通过对CHRM3基因突变小鼠的研究, Poulin等人认为CHRM3突变小鼠的认知功能受损是由CHRM3不能正常磷酸化导致的。CHRM3受体磷酸化发生在第384号丝氨酸位点上, 当编码该位点氨基酸的基因突变后, CHRM3无法正常磷酸化, 影响了β-arrestin与CHRM3的结合过程, 导致受体内在化过程受阻, 最终阻断了神经信号通路的信号传递过程, 小鼠表现出认知能力受损的特征。为了进一步了解CHRM3如何影响小鼠的学习记忆能力, 研究者测定了小鼠海马神经元中c-fos基因的表达水平。在环境联系性学习过程中, 突触后神经元兴奋产生长时程增强(long term potentiation, LTP)激活c-fos基因。c-fos基因编码的磷酸蛋白可作为转录因子与DNA结合, 促进或抑制相关基因的表达, 从而把由外界刺激所诱发的短暂的细胞内信息与由基因改变所产生的突触可塑性过程偶联起来, 一旦再次接受该环境刺激时, c-fos基因的表达水平会迅速增加, 因此诱导c-fos mRNA的表达可能是形成长时记忆的必要条件(Beck & Fibiger, 1995; Tischmeyer, Kaczmarek, Strauss, Jork, & Matthies, 1990)。Poulin等人的结果显示CHRM3突变小鼠海马和齿状回内c-fos基因表达水平显著低于野生型小鼠。Rosethorne、Nahorski和Challiss (2008)也曾发现CHRM3对c-fos表达起着调节作用:CHRM3可以促进CREB (cAMP response-element binding protein)磷酸化, 而CREB磷酸化能够诱导c-fos基因表达, 因此激活CHRM3可以提高c-fos的表达水平。值得注意的是, CREB在神经元发育、突触可塑性建立、学习记忆过程中起着重要的调节功能(Silva, Kogan, Frankland, & Kida, 1998; Lonze & Ginty, 2002; Carlezon, Duman, & Nestler, 2005)。综合以上研究推测, CHRM3突变小鼠学习记忆能力较低的原因可能是由于学习记忆相关神经元内依赖Gq-PLC的钙离子信号通路信号传递受阻抑制了CREB磷酸化, 进而抑制了c-fos基因启动应对环境刺激反应的下游基因的表达, 因此无法激活与学习记忆相关神经元, 特定脑区功能受损, 最终表现为个体学习记忆能力较低, 无法在短时间内习得应对环境刺激的反应。除此之外, 在Karvat和Kimchi (2014)的研究中还发现, 向BTBR小鼠背内侧纹状体注射乙酰胆碱酯酶抑制剂后可以有效改善小鼠的学习能力缺陷的问题(Karvat & Kimchi, 2014)。由此可见, 在后续研究中可以通过向BTBR小鼠的海马或背内侧纹状体注射CHRM3特异性激动剂, 观察小鼠是否表现出学习记忆能力变化, 并测定c-fos表达量来进一步探究CHRM3基因在孤独症患者认知活动中的作用。
当前关于认知功能机制的研究大多集中于边缘系统, CHRM3突变的孤独症患者的认知功能受损主要被认为与海马功能异常有关, 但对此也有不同的观点, 有研究者认为CHRM3介导的信号传递过程可能是小脑浦肯野细胞突触形成的主要机制(Rinaldo & Hansel, 2013), 因此CHRM3突变的孤独症患者的认知障碍或许是由小脑功能异常所致, 这还需要在今后的研究中进一步探讨。
4.3 CHRM3异常与孤独症生长发育迟缓
研究发现, 孤独症患者中出现生长发育迟缓问题的比例较高(Haglund & Kallen, 2010), 因此有学者提出生长发育迟缓可能是导致孤独症发生的中介因素之一(Haglund & Kallen, 2010)。在已报道的CHRM3基因异常的临床案例中, 患者均出现了发育迟缓的症状。
动物模型研究发现CHRM3敲除小鼠会出现体重减轻, 摄食量减少、血清内瘦素和胰岛素水平显著降低等一系列生长发育迟缓的特征(Yamada et al., 2001; Matsui et al., 2000; Meyer, Zhu, Miller, & Roghair, 2014), 这与Perrone等人(2012)和Shimojima等人(2012)报道的患者的临床表现相似。研究人员发现在野生型小鼠脑内, CHRM3主要分布在下丘脑, 而CHRM3敲除小鼠下丘脑内CHRM3数量与野生型小鼠相比下降了近50%, 同时免疫组化研究显示小鼠下丘脑内黑色素聚集激素(melanin- concentrating hormone, MCH)的表达水平也显著低于野生型小鼠(Yamada et al., 2001)。已有研究证实MCH对于调控摄食和体重变化具有重要作用(Qu et al., 1996), 且CHRM3与MCH被证实在外侧下丘脑细胞内共表达, 因此Ymada等人推测在有关饮食调节的信号通路中, 瘦素和胰岛素作为上游的信号因子刺激下丘脑弓形核, 激活MCH细胞, 从而激活了下丘脑信号通路, 开启信号转导过程。在该信号通路下游的外侧下丘脑内, CHRM3通过控制MCH细胞分泌MCH从而调控个体的摄食行为, 即当外侧下丘脑内的MCH细胞接收到乙酰胆碱信号刺激后, CHRM3被激活, MCH释放量迅速提高, 个体出现摄食行为。因此在CHRM3敲除小鼠体内, CHRM3缺失导致MCH细胞无法被激活释放MCH, 小鼠摄食量下降, 进而表现出体重减轻等发育迟缓的问题症状。
由于瘦素是激活下丘脑饮食调节信号通路的主要因子, 因此瘦素含量降低也会导致个体出现生长发育迟缓的症状(Meyer et al.,2014)。研究发现, 婴儿期瘦素缺失将导致发育迟缓的小鼠在成年期出现运动能力降低、社交兴趣丧失、认知能力受损、以及杏仁核体积增大等孤独症样的异常特征(Meyer et al., 2014)。因此婴儿期个体瘦素水平降低可能与孤独症的发生有关。结合在Ymada等人的研究中CHRM3敲除小鼠血清内瘦素含量显著降低这一结果, 推测瘦素含量下降与CHRM3基因表达水平降低有关, 早期营养不足可能是后期行为问题出现的原因之一, 即CHRM3缺失会降低个体的摄食行为, 在一定程度上影响身体生长和脑的发育过程, 最终导致问题行为出现。
另外, 免疫组化研究证实小鼠唾液腺上2/3的M型受体为CHRM3受体, 说明CHRM3对于调控唾液分泌也具有重要作用(Matsui et al., 2000; Bymaster et al., 2003), 因此CHRM3突变的生长发育迟缓小鼠出现进食障碍有可能是由于唾液分泌过程异常引起的食物消化功能受损所致。以上研究表明CHRM3与生长发育之间有着紧密的联系, 一方面CHRM3可以通过调节摄食行为来影响生长发育, 另一方面可以通过调节消化能力影响生长发育。
5 总结与展望
作为G蛋白偶联受体家族一员, CHRM3介导Gq-PLC信号通路参与突触信号传递, 对于调控细胞增殖、代谢、细胞骨架建立和突触可塑性形成具有重要作用。由于突触依赖性的神经元信号传导是学习、记忆等高级心理活动的生理基础, 因此CHRM3可能与人的认知能力发展以及社会化等发育过程密切相关。
临床案例和动物模型研究均发现改变CHRM3功能会引发动物出现认知缺陷以及刻板行为等孤独症特征(见图2)。抑制CHRM3基因的表达将会影响受体磷酸化过程, 降低海马、杏仁核、嗅球等组织中神经元的活跃水平, 进而导致一系列异常行为特征出现。而过表达CHRM3会导致海马内兴奋性神经元被过度激活, 也会影响孤独症样行为出现, 因此无论CHRM3所介导的神经信号通路被抑制或是增强, 一旦神经系统内环境稳态被破坏都有可能引发孤独症的发生。鉴于此, 控制Gq-PLC信号通路活动水平适中对于特定行为的发展有重要作用。但选择哪一项指标作为衡定信号通路适中的标准, 尚有待今后的深入研究。除此之外, 当孤独症高风险基因发生突变时, CHRM3的表达也会受到影响(Forrest, Waite, Martin-Rendon, & Blake, 2013; Chan et al., 2015)。另外在对孤独症患者家系全基因组检测中, 发现了一个CHRM3下游分子PLC家族成员(磷酯酶)的编码基因存在新生突变, 这暗示CHRM3及其所调控信号通路对孤独症发生发展有重要影响。但是目前有关CHRM3基因突变在孤独症发生发展中的作用以及在脑发育过程中的机制还有待进一步探讨。
图2

在接下来的研究中, 可以在建立小鼠动物模型的基础上, 通过检测基因分子水平变化、细胞组织器官发育分化、形态差异以及分析行为特征来研究CHRM3基因在神经系统发育中的作用, 及其对孤独症发生的影响。另外, 关注CHRM3所介导的Gq-PLC信号通路在孤独症发生中的作用, 可为孤独症的基因靶向干预提供新的思路和方法, 为教育方案的制定提供科学的帮助和指导。
参考文献
Remote control of neuronal activity in transgenic mice expressing evolved g protein-coupled receptors
DOI:10.1016/j.neuron.2009.06.014
URL
PMID:19607790
[本文引用: 2]

Examining the behavioral consequences of selective CNS neuronal activation is a powerful tool for elucidating mammalian brain function in health and disease. Newly developed genetic, pharmacological, and optical tools allow activation of neurons with exquisite spatiotemporal resolution; however, the inaccessibility to light of widely distributed neuronal populations and the invasiveness required for activation by light or infused ligands limit the utility of these methods. To overcome these barriers, we created transgenic mice expressing an evolved G protein-coupled receptor (hM3Dq) selectively activated by the pharmacologically inert, orally bioavailable drug clozapine-N-oxide (CNO). Here, we expressed hM3Dq in forebrain principal neurons. Local field potential and single-neuron recordings revealed that peripheral administration of CNO activated hippocampal neurons selectively in hM3Dq-expressing mice. Behavioral correlates of neuronal activation included increased locomotion, stereotypy, and limbic seizures. These results demonstrate a powerful chemical-genetic tool for remotely controlling the activity of discrete populations of neurons in vivo.
Oxotremorine treatment reduces repetitive behaviors in btbr t+ tf/j mice
DOI:10.3389/fnsyn.2014.00017
URL
PMID:25165445
[本文引用: 2]
Repetitive behaviors with restricted interests is one of the core criteria for the diagnosis of autism spectrum disorder (ASD). Current pharmacotherapies that target the dopaminergic or serotonergic systems have limited effectiveness in treating repetitive behaviors. Previous research has demonstrated that administration of muscarinic cholinergic receptor (mAChR) antagonists can exacerbate motor stereotypies while mAChR agonists reduce stereotypies. The present study determined whether the mAChR agonist, oxotremorine affected repetitive behaviors in the BTBR T+ tf/J (BTBR) mouse model of autism. To test the effects of oxotremorine on repetitive behaviors, marble burying and grooming behavior were measured in BTBR mice and compared to that in C57BL/6J (B6) mice. The effects of oxotremorine on locomotor activity was also measured. Thirty minutes before each test, mice received an intraperitoneal (ip) injection of saline, 0.001 mg or 0.01 mg of oxotremorine methiodide. Saline- treated BTBR mice exhibited increased marble burying and self-grooming behavior compared to that of saline-treated B6 mice. Oxotremorine significantly reduced marble burying and self-grooming behavior in BTBR mice, but had no significant effect in B6 mice. In addition, oxotremorine did not affect locomotor activity in BTBR mice, but significantly reduced locomotor activity in B6 mice at the 0.01 mg dose. These findings demonstrate that activation of mAChRs reduces repetitive behavior in the BTBR mouse and suggest that treatment with a mAChR agonist may be effective in reducing repetitive behaviors in ASD.
Autism as a strongly genetic disorder: Evidence from a British twin study
DOI:10.1017/S0033291700028099
URL
PMID:7792363132
[本文引用: 1]
Two previous epidemiological studies of autistic twins suggested that autism was predominantly genetically determined, although the findings with regard to a broader phenotype of cognitive, and possibly social, abnormalities were contradictory. Obstetric and perinatal hazards were also invoked as environmentally determined aetiological factors. The first British twin sample has been re-examined and a second total population sample of autistic twins recruited. In the combined sample 60% of monozygotic (MZ) pairs were concordant for autismversusno dizygotic (DZ) pairs; 92% of MZ pairs were concordant for a broader spectrum of related cognitive or social abnormalitiesversus10% of DZ pairs. The findings indicate that autism is under a high degree of genetic control and suggest the involvement of multiple genetic loci. Obstetric hazards usually appear to be consequences of genetically influenced abnormal development, rather than independent aetiological factors. Few new cases had possible medical aetiologies, refuting claims that recognized disorders are common aetiological influences.
Conditioned fear-induced changes in behavior and in the expression of the immediate early gene c-fos: With and without diazepam pretreatment
DOI:10.1051/jphyscol:1990171
URL
PMID:7823174
[本文引用: 1]
Abstract The synthesis of Fos, the protein product of the immediate early gene c-fos, was used to map metabolically some of the neural substrates of conditioned fear in the rat. Analysis of the behaviors emitted by the rats during the test session provided strong evidence that the conditioning procedure was effective. Exposure to the environment in which they had previously received footshock significantly increased the number of Fos-like immunoreactive neurons in nearly 50 brain regions, both cortical and subcortical. Among the structures showing the most dramatic increases in fear-induced c-fos expression were the cingulate, piriform, infralimbic, and retrosplenial cortices, the anterior olfactory nucleus, claustrum, endopiriform nucleus, nucleus accumbens shell, lateral septal nucleus, various amygdalar nuclei, paraventricular thalamic nucleus, ventral lateral geniculate nucleus, the ventromedial, lateral, and dorsal hypothalamic nuclei, the ventral tegmental area, and the supramammillary area. These data demonstrate that a relatively simple classical conditioning procedure activates a large number of widely dispersed cortical and subcortical structures. Some of the structures showing increased c-fos expression have important autonomic functions and may therefore have reflected centrally mediated changes in blood pressure and respiration produced by the anxiogenic stimuli. In a second experiment, the effects of pretreatment with the anxiolytic drug diazepam (2.5, 5.0, or 10 mg/kg) were evaluated. The benzodiazepine produced dose-related decreases in the frequency of crouching (freezing) elicited by the aversively conditioned contextual cues. Diazepam also produced dose-related decreases in conditioned stress-induced c-fos expression in all but one structure, the effects being statistically significant in 38 of 60 sampled structures. Diazepam dose dependently increased fear-induced c-fos expression in the central nucleus of the amygdala. There was considerable regional variability with respect to sensitivity to diazepam, the retrosplenial cortex and the supramammillary area being the only two structures to show decreases after the lowest dose of diazepam. In contrast, the entorhinal cortex, nucleus accumbens core, ventromedial and posterior hypothalamic nuclei, median raphe, and locus coeruleus were particularly resistant to diazepam, all failing to show statistically significant decreases in conditioned fear-induced c-fos expression even at the highest dose. The extent to which diazepam decreased conditioned stress-induced c-fos expression was unrelated to previous estimates of benzodiazepine receptor density in the sampled structures.
Cholinergic enhancement modulates neural correlates of selective attention and emotional processing
Disruptive CHD8 mutations define a subtype of autism early in development
DOI:10.1016/j.cell.2014.06.017
URL
PMID:24998929
[本文引用: 2]
Identification of patients all harboring mutations in the ASD risk gene CHD8 revealed common phenotypes among them, indicating that CHD8 disruptions define a distinct ASD subtype characterized, in part, by macrocephaly and impaired gastrointestinal function.
Assessing autism-like behavior in mice: Variations in social interactions among inbred strains
DOI:10.1016/j.bbr.2006.09.007
URL
PMID:1831820
[本文引用: 1]
Autism is a pervasive developmental disorder, with characteristics including impairments in reciprocal social interaction, impaired communication, and repetitive/stereotyped behaviors. Despite decades of research, the etiology of autism remains elusive. Thus, it is important that we pursue all avenues, in attempting to understand this complicated disorder. One such avenue is the development of animal models. While autism may be uniquely human, there are behavioral characteristics of the disorder that can be established in animal models. Evidence supports a genetic component for this disorder, and over the past few decades the mouse has been a highly valuable tool for the elucidation of pathways involved in many human disorders (e.g., Huntington's disease). As a first step toward establishing a mouse model of autism, we studied same-sex social behavior in a number of inbred mouse strains. In Study 1, we examined intra-strain social behavior of male pairs after one mouse had 15 min prior exposure to the testing chamber. In Study 2, we evaluated intra-strain and inter-strain social behavior when both mice were naive to the testing chamber. The amount and type of social behavior seen differed between these studies, but overall there were general inbred strain differences in social behavior. Some strains were highly social, e.g., FVB/NJ, while others displayed low levels of social behavior (e.g., A/J, BTBR T + tf/J). These strains may be useful in future genetic studies to determine specific genes involved in mouse social behavior, the findings of which should in turn help us to determine some of the genes involved in human social behavior and its disorders (e.g., autism).
Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies
High-resolution chromosome ideogram representation of currently recognized genes for autism spectrum disorders
DOI:10.3390/ijms16036464
URL
PMID:25803107
[本文引用: 1]
Recently, autism-related research has focused on the identification of various genes and disturbed pathways causing the genetically heterogeneous group of autism spectrum disorders (ASD). The list of autism-related genes has significantly increased due to better awareness with advances in genetic technology and expanding searchable genomic databases. We compiled a master list of known and clinically relevant autism spectrum disorder genes identified with supporting evidence from peer-reviewed medical literature sources by searching key words related to autism and genetics and from authoritative autism-related public access websites, such as the Simons Foundation Autism Research Institute autism genomic database dedicated to gene discovery and characterization. Our list consists of 792 genes arranged in alphabetical order in tabular form with gene symbols placed on high-resolution human chromosome ideograms, thereby enabling clinical and laboratory geneticists and genetic counsellors to access convenient visual images of the location and distribution of ASD genes. Meaningful correlations of the observed phenotype in patients with suspected/confirmed ASD gene(s) at the chromosome region or breakpoint band site can be made to inform diagnosis and gene-based personalized care and provide genetic counselling for families.
Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity
DOI:10.1086/320588
URL
PMID:11353400
[本文引用: 1]
Although there is considerable evidence for a strong genetic component to idiopathic autism, several genomewide screens for susceptibility genes have been performed with limited concordance of linked loci, reflecting either numerous genes of weak effect and/or sample heterogeneity. Because decreasing sample heterogeneity would increase the power to identify genes, the effect on evidence for linkage of restricting a sample of autism-affected relative pairs to those with delayed onset (at age >36 mo) of phrase speech (PSD, for phrase speech delay) was studied. In the second stage of a two-stage genome screen for susceptibility loci involving 95 families with two or more individuals with autism or related disorders, a maximal multipoint heterogeneity LOD score (HLOD) of 1.96 and a maximal multipoint nonparametric linkage (NPL) score of 2.39 was seen on chromosome 2q. Restricting the analysis to the subset of families ( n=49) with two or more individuals having a narrow diagnosis of autism and PSD generated a maximal multipoint HLOD score of 2.99 and an NPL score of 3.32. The increased scores in the restricted sample, together with evidence for heterogeneity in the entire sample, indicate that the restricted sample comprises a population that is more genetically homogeneous, which could therefore increase the likelihood of positional cloning of susceptibility loci.
Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine- induced seizure activity
The many faces of CREB
DOI:10.1016/j.tins.2005.06.005
URL
PMID:15982754
[本文引用: 1]
The transcription factor CREB is best known for its involvement in learning and memory. However, emerging evidence suggests that CREB activity has very different roles – sometimes beneficial, sometimes detrimental – depending on the brain region involved. Induction of CREB in the hippocampus by antidepressant treatments could contribute to their therapeutic efficacy. By contrast, activation of CREB in the nucleus accumbens and several other regions by drugs of abuse or stress mediates certain aspects of drug addiction, and depressive and anxiety-like behaviors. These complexities suggest that strategies that exploit regional differences in upstream factors or that target specific CREB-regulated genes, rather than CREB itself, could make a promising contribution to the treatment of neuropsychiatric conditions.
Transcriptional profiling of MEF2-regulated genes in human neural progenitor cells derived from embryonic stem cells
DOI:10.1016/j.gdata.2014.10.022
URL
PMID:4255278
[本文引用: 1]
The myocyte enhancer factor 2 (MEF2) family of transcription factors is highly expressed in the brain and constitutes a key determinant of neuronal survival, differentiation, and synaptic plasticity. However, genome-wide transcriptional profiling of MEF2-regulated genes has not yet been fully elucidated, particularly at the neural stem cell stage. Here we report the results of microarray analysis comparing mRNAs isolated from human neural progenitor/stem cells (hNPCs) derived from embryonic stem cells expressing a control vector versus progenitors expressing a constitutively-active form of MEF2 (MEF2CA), which increases MEF2 activity. Microarray experiments were performed using the Illumina Human HT-12V4.0 expression beadchip (GEO#: GSE57184). By comparing vector-control cells to MEF2CA cells, microarray analysis identified 1880 unique genes that were differentially expressed. Among these genes, 1121 genes were up-regulated and 759 genes were down-regulated. Our results provide a valuable resource for identifying transcriptional targets of MEF2 in hNPCs.
Sapap3 deletion anomalously activates short-term endocannabinoid-mediated synaptic plasticity
DOI:10.1523/JNEUROSCI.1701-11.2011
URL
PMID:21715621
[本文引用: 2]
Retrograde synaptic signaling by endocannabinoids (eCBs) is a widespread mechanism for activity-dependent inhibition of synaptic strength in the brain. Although prevalent, the conditions for eliciting eCB-mediated synaptic depression vary among brain circuits. As yet, relatively little is known about the molecular mechanisms underlying this variation, although the initial signaling events are likely dictated by postsynaptic proteins. SAP90/PSD-95-associated proteins (SAPAPs) are a family of postsynaptic proteins unique to excitatory synapses. Using Sapap3 knock-out (KO) mice, we find that, in the absence of SAPAP3, striatal medium spiny neuron (MSN) excitatory synapses exhibit eCB-mediated synaptic depression under conditions that do not normally activate this process. The anomalous synaptic plasticity requires type 5 metabotropic glutamate receptors (mGluR5s), which we find are dysregulated in Sapap3 KO MSNs. Both surface expression and activity of mGluR5s are increased in Sapap3 KO MSNs, suggesting that enhanced mGluR5 activity may drive the anomalous synaptic plasticity. In direct support of this possibility, we find that, in wild-type (WT) MSNs, pharmacological enhancement of mGluR5 by a positive allosteric modulator is sufficient to reproduce the increased synaptic depression seen in Sapap3 KO MSNs. The same pharmacologic treatment, however, fails to elicit further depression in KO MSNs. Under conditions that are sufficient to engage eCB-mediated synaptic depression in WT MSNs, Sapap3 deletion does not alter the magnitude of the response. These results identify a role for SAPAP3 in the regulation of postsynaptic mGluRs and eCB-mediated synaptic plasticity. SAPAPs, through their effect on mGluR activity, may serve as regulatory molecules gating the threshold for inducing eCB-mediated synaptic plasticity.
Meta-analysis of gene expression in autism spectrum disorder
DOI:10.1002/aur.1475
URL
PMID:25720351
[本文引用: 1]
Autism spectrum disorders (ASD) are clinically heterogeneous and biologically complex. In general it remains unclear, what biological factors lead to changes in the brains of autistic individuals. A considerable number of transcriptome analyses have been performed in attempts to address this question, but their findings lack a clear consensus. As a result, each of these individual studies has not led to any significant advance in understanding the autistic phenotype as a whole. Here, we report a meta-analysis of more than 1000 microarrays across twelve independent studies on expression changes in ASD compared to unaffected individuals, in both blood and brain tissues. We identified a number of known and novel genes that are consistently differentially expressed across three studies of the brain (71 samples in total). A subset of the highly ranked genes is suggestive of effects on mitochondrial function. In blood, consistent changes were more difficult to identify, despite individual studies tending to exhibit larger effects than the brain studies. Our results are the strongest evidence to date of a common transcriptome signature in the brains of individuals with ASD.
Prevalence and characteristics of autism spectrum disorder among children aged 8 years--Autism and developmental disabilities monitoring network, 11 sites, United States, 2012
Patterns of autobiographical memory in adults with autism spectrum disorder
DOI:10.1007/s10803-012-1459-2
URL
PMID:22322581
[本文引用: 1]
Two studies are presented that explored the effects of experimental manipulations on the quality and accessibility of autobiographical in adults with (), relative to a typical comparison group matched for age, gender and IQ. Both studies found that the adults with generated fewer specific than the comparison group, and took significantly longer to do so. Despite this, experimental manipulations affected two indices of autobiographical (specificity and retrieval latency) similarly in both groups. These results suggest that adults with experience a quantitative reduction in the speed and specificity of autobiographical retrieval, but that when they do retrieve these , they do so in a way that is qualitatively similar to that of typical adults.
Factor analysis of restricted and repetitive behaviors in autism using the autism diagnostic interview-r
DOI:10.1023/A:1025321707947
URL
PMID:14518620
[本文引用: 1]
The current study examined the factor structure of restricted and repetitive behaviors (RRB) in children with autism. Factor extraction procedures of 12 items from the Autism Diagnostic Interview-Revised ( ADI-R ) were applied in N = 207 individuals with autism. Two interpretable factors were identified: Factor 1— repetitive sensory motor actions and Factor 2— resistance to change . There was a significant negative correlation between an index of level of adaptive functioning and Factor 1. Intraclass correlations were not significant for either factor in a subset of families with two or more siblings with autism (multiplex). No differences in scores were apparent for either factor when multiplex families and families containing only one affected individual with autism (singleton) were compared. RRB in autism are represented by two distinct factors which may reflect two separate groups within autism. Defining subgroups within autism will allow for reduction of clinical heterogeneity and enhance our ability to dissect the genetic etiology of this complex disorder.
Autistic-like traits and cerebellar dysfunction in Purkinje cell PTEN knock- out mice
DOI:10.1038/npp.2015.339
URL
PMID:4832032
[本文引用: 1]
Abstract Autism spectrum disorders (ASDs) are neurodevelopmental disorders characterized by impaired social interaction, isolated areas of interest, and insistence on sameness. Mutations in Phosphatase and tensin homolog missing on chromosome 10 (PTEN) have been reported in individuals with ASDs. Recent evidence highlights a crucial role of the cerebellum in the etiopathogenesis of ASDs. In the present study we analyzed the specific contribution of cerebellar Purkinje cell (PC) PTEN loss to these disorders. Using the Cre-loxP recombination system, we generated conditional knockout mice in which PTEN inactivation was induced specifically in PCs. We investigated PC morphology and physiology as well as sociability, repetitive behavior, motor learning, and cognitive inflexibility of adult PC PTEN-mutant mice. Loss of PTEN in PCs results in autistic-like traits, including impaired sociability, repetitive behavior and deficits in motor learning. Mutant PCs appear hypertrophic and show structural abnormalities in dendrites and axons, decreased excitability, disrupted parallel fiber and climbing fiber synapses and late-onset cell death. Our results unveil new roles of PTEN in PC function and provide the first evidence of a link between the loss of PTEN in PCs and the genesis of ASD-like traits.
Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system
Synaptic, transcriptional and chromatin genes disrupted in autism
DOI:10.1038/nature13772
URL
PMID:25363760
[本文引用: 1]
Abstract The genetic architecture of autism spectrum disorder involves the interplay of common and rare variants and their impact on hundreds of genes. Using exome sequencing, here we show that analysis of rare coding variation in 3,871 autism cases and 9,937 ancestry-matched or parental controls implicates 22 autosomal genes at a false discovery rate (FDR) < 0.05, plus a set of 107 autosomal genes strongly enriched for those likely to affect risk (FDR < 0.30). These 107 genes, which show unusual evolutionary constraint against mutations, incur de novo loss-of-function mutations in over 5% of autistic subjects. Many of the genes implicated encode proteins for synaptic formation, transcriptional regulation and chromatin-remodelling pathways. These include voltage-gated ion channels regulating the propagation of action potentials, pacemaking and excitability-transcription coupling, as well as histone-modifying enzymes and chromatin remodellers-most prominently those that mediate post-translational lysine methylation/demethylation modifications of histones.
Cholinergic interneurons in the Q140 knockin mouse model of Huntington's disease: Reductions in dendritic branching and thalamostriatal input
Cholinergic abnormalities in autism: Is there a rationale for selective nicotinic agonist interventions?
DOI:10.1097/WNF.0b013e3181d6f7ad
URL
PMID:20190638
[本文引用: 1]
The core dysfunctions of , which include , , and not otherwise specified, include deficits in socialization and communication and a need for the preservation of "sameness;" and are common comorbidities. Data suggest that pathological involvement of cholinergic nuclei and altered expression of receptors, particularly nicotinic receptors, occur in brain of persons with . However, many of these studies involved postmortem tissue from small samples of primarily adult persons. Thus, the findings may reflect compensatory changes and may relate more closely to and the confounding effects of seizures and medications, as opposed to the core dysfunctions of . Nonetheless, because of the roles played by receptors in general, and nicotinic receptors in particular, in normal processes of attention, , and , selective cholinergic interventions should be explored for possible therapeutic effects. Additionally, there are electrophysiological data that complement the clinical observations of frequent comorbid in these patients, suggesting a disturbance in the balance of excitatory and inhibitory tone in the brains of persons with . Conceivably, because the alpha7 nicotinic receptor is located on the surface of inhibitory neurons, selective stimulation of this receptor would promote 's release and restore diminished inhibitory tone. The of and partial for nicotinic receptors and positive allosteric modulators that enhance the efficiency of coupling between the of and channel opening should facilitate consideration of clinical trials.
Genetic evidence for role of integration of fast and slow neurotransmission in schizophrenia
DOI:10.1038/mp.2017.33
URL
PMID:28348379
[本文引用: 1]
The most recent genome-wide association studies (GWAS) of schizophrenia (SCZ) identified hundreds of risk variants potentially implicated in the disease. Further, novel statistical methodology designed for polygenic architecture revealed more potential risk variants. This can provide a link between individual genetic factors and the mechanistic underpinnings of SCZ. Intriguingly, a large number of genes coding for ionotropic and metabotropic receptors for various neurotransmitters—glutamate, γ-aminobutyric acid (GABA), dopamine, serotonin, acetylcholine and opioids—and numerous ion channels were associated with SCZ. Here, we review these findings from the standpoint of classical neurobiological knowledge of neuronal synaptic transmission and regulation of electrical excitability. We show that a substantial proportion of the identified genes are involved in intracellular cascades known to integrate ‘slow’ (G-protein-coupled receptors) and ‘fast’ (ionotropic receptors) neurotransmission converging on the protein DARPP-32. Inspection of the Human Brain Transcriptome Project database confirms that that these genes are indeed expressed in the brain, with the expression profile following specific developmental trajectories, underscoring their relevance to brain organization and function. These findings extend the existing pathophysiology hypothesis by suggesting a unifying role of dysregulation in neuronal excitability and synaptic integration in SCZ. This emergent model supports the concept of SCZ as an ‘associative’ disorder—a breakdown in the communication across different slow and fast neurotransmitter systems through intracellular signaling pathways—and may unify a number of currently competing hypotheses of SCZ pathophysiology.
Nicotinic ACh receptors as therapeutic targets in CNS disorders
DOI:10.1016/j.tips.2014.12.002
URL
PMID:4324614
[本文引用: 1]
The neurotransmitter acetylcholine (ACh) can regulate neuronal excitability by acting on the cys-loop cation-conducting ligand-gated nicotinic ACh receptor (nAChR) channels. These receptors are widely distributed throughout the central nervous system (CNS), being expressed on neurons and non-neuronal cells, where they participate in a variety of physiological responses such as anxiety, the central processing of pain, food intake, nicotine seeking behavior, and cognitive functions. In the mammalian brain, nine different subunits have been found thus far, which assemble into pentameric complexes with much subunit diversity; however, the 7 and 4 2 subtypes predominate in the CNS. Neuronal nAChR dysfunction is involved in the pathophysiology of many neurological disorders. Here we will briefly discuss the functional makeup and expression of the nAChRs in mammalian brain, and their role as targets in neurodegenerative diseases (in particular Alzheimer's disease, AD), neurodevelopmental disorders (in particular autism and schizophrenia), and neuropathic pain.
Early- and late-born parvalbumin basket cell subpopulations exhibiting distinct regulation and roles in learning
DOI:10.1016/j.neuron.2015.01.011
URL
PMID:25695271
[本文引用: 1]
Brain networks can support learning by promoting acquisition of task-relevant information or by adhering to validated rules. This study shows that these opposite learning requirements are implemented through previously unrecognized parvalbumin Basket cell subpopulations specifically regulated through inhibition or excitation.
Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders
DOI:10.1038/ng1933
URL
PMID:2017173049
[本文引用: 1]
Abstract SHANK3 (also known as ProSAP2) regulates the structural organization of dendritic spines and is a binding partner of neuroligins; genes encoding neuroligins are mutated in autism and Asperger syndrome. Here, we report that a mutation of a single copy of SHANK3 on chromosome 22q13 can result in language and/or social communication disorders. These mutations concern only a small number of individuals, but they shed light on one gene dosage-sensitive synaptic pathway that is involved in autism spectrum disorders.
Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation
DOI:10.1371/journal.pone.0073169
URL
PMID:3751932
[本文引用: 1]
Haploinsufficiency ofTCF4causes Pitt-Hopkins syndrome (PTHS): a severe form of mental retardation with phenotypic similarities to Angelman, Mowat-Wilson and Rett syndromes. Genome-wide association studies have also found that common variants inTCF4are associated with an increased risk of schizophrenia. Although TCF4 is transcription factor, little is known about TCF4-regulated processes in the brain. In this study we used genome-wide expression profiling to determine the effects of acute TCF4 knockdown on gene expression in SH-SY5Y neuroblastoma cells. We identified 1204 gene expression changes (494 upregulated, 710 downregulated) in TCF4 knockdown cells. Pathway and enrichment analysis on the differentially expressed genes in TCF4-knockdown cells identified an over-representation of genes involved in TGF- signaling, epithelial to mesenchymal transition (EMT) and apoptosis. Among the most significantly differentially expressed genes were the EMT regulators,SNAI2andDEC1and the proneural genes,NEUROG2andASCL1. Altered expression of several mental retardation genes such asUBE3A(Angelman Syndrome),ZEB2(Mowat-Wilson Syndrome) andMEF2Cwas also found in TCF4-depleted cells. These data suggest that TCF4 regulates a number of convergent signaling pathways involved in cell differentiation and survival in addition to a subset of clinically important mental retardation genes.
Gray and white matter brain chemistry in young children with autism
Rare structural variation of synapse and neurotransmission genes in autism
DOI:10.1038/mp.2011.10
URL
PMID:21358714
[本文引用: 1]
Abstract Autism spectrum disorders (ASDs) comprise a constellation of highly heritable neuropsychiatric disorders. Genome-wide studies of autistic individuals have implicated numerous minor risk alleles but few common variants, suggesting a complex genetic model with many contributing loci. To assess commonality of biological function among rare risk alleles, we compared functional knowledge of genes overlapping inherited structural variants in idiopathic ASD subjects relative to healthy controls. In this study we show that biological processes associated with synapse function and neurotransmission are significantly enriched, with replication, in ASD subjects versus controls. Analysis of phenotypes observed for mouse models of copy-variant genes established significant and replicated enrichment of observable phenotypes consistent with ASD behaviors. Most functional terms retained significance after excluding previously reported ASD loci. These results implicate several new variants that involve synaptic function and glutamatergic signaling processes as important contributors of ASD pathophysiology and suggest a sizable pool of additional potential ASD risk loci.
An AUTS2-Polycomb complex activates gene expression in the CNS
DOI:10.1038/nature13921
URL
PMID:25519132
[本文引用: 1]
Abstract Naturally occurring variations of Polycomb repressive complex 1 (PRC1) comprise a core assembly of Polycomb group proteins and additional factors that include, surprisingly, autism susceptibility candidate 2 (AUTS2). Although AUTS2 is often disrupted in patients with neuronal disorders, the mechanism underlying the pathogenesis is unclear. We investigated the role of AUTS2 as part of a previously identified PRC1 complex (PRC1-AUTS2), and in the context of neurodevelopment. In contrast to the canonical role of PRC1 in gene repression, PRC1-AUTS2 activates transcription. Biochemical studies demonstrate that the CK2 component of PRC1-AUTS2 neutralizes PRC1 repressive activity, whereas AUTS2-mediated recruitment of P300 leads to gene activation. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) demonstrated that AUTS2 regulates neuronal gene expression through promoter association. Conditional targeting of Auts2 in the mouse central nervous system (CNS) leads to various developmental defects. These findings reveal a natural means of subverting PRC1 activity, linking key epigenetic modulators with neuronal functions and diseases.
Most genetic risk for autism resides with common variation
DOI:10.1038/ng.3039
URL
PMID:25038753
[本文引用: 1]
Abstract A key component of genetic architecture is the allelic spectrum influencing trait variability. For autism spectrum disorder (herein termed autism), the nature of the allelic spectrum is uncertain. Individual risk-associated genes have been identified from rare variation, especially de novo mutations. From this evidence, one might conclude that rare variation dominates the allelic spectrum in autism, yet recent studies show that common variation, individually of small effect, has substantial impact en masse. At issue is how much of an impact relative to rare variation this common variation has. Using a unique epidemiological sample from Sweden, new methods that distinguish total narrow-sense heritability from that due to common variation and synthesis of results from other studies, we reach several conclusions about autism's genetic architecture: its narrow-sense heritability is 52.4%, with most due to common variation, and rare de novo mutations contribute substantially to individual liability, yet their contribution to variance in liability, 2.6%, is modest compared to that for heritable variation.
Risk factors for autism and Asperger syndrome. Perinatal factors and migration
DOI:10.1177/1362361309353614
URL
PMID:20923887
[本文引用: 2]
Using the Swedish Medical Birth Registry (MBR), obstetrical and demographic information was retrieved for 250 children with autism or Asperger syndrome who were born in Malmoe, Sweden, and enrolled at the local Child and Youth Habilitation Center. The reference group consisted of all children born in Malmoe during 1980-2005. Obstetric sub-optimality (prematurity, low Apgar scores, growth restriction, or macrosomia) was positively associated with autism but not with Asperger syndrome. Maternal birth outside the Nordic countries was positively associated with autism (adjusted OR: 2.2; 95% CI: 1.6-3.1) and negatively associated with Asperger syndrome (OR: 0.6; 95% CI: 0.3-0.97). The highest risk estimate for autism was found among children to women who were born in sub-Saharan Africa (OR: 7.3), or in East Asia (OR: 3.4).
Time to give up on a single explanation for autism
DOI:10.1038/nn1770
URL
PMID:17001340
[本文引用: 1]
Abstract We argue that there will be no single (genetic or cognitive) cause for the diverse symptoms defining autism. We present recent evidence of behavioral fractionation of social impairment, communication difficulties and rigid and repetitive behaviors. Twin data suggest largely nonoverlapping genes acting on each of these traits. At the cognitive level, too, attempts at a single explanation for the symptoms of autism have failed. Implications for research and treatment are discussed.
Retrospective study of quetiapine in children and adolescents with pervasive developmental disorders
DOI:10.1007/s10803-005-3306-1
URL
PMID:16119479
[本文引用: 1]
A retrospective study was conducted in a clinic specialized in treating individuals with developmental disabilities to examine the effectiveness and tolerability of quetiapine in children and adolescents with pervasive developmental disorders. Ten consecutive outpatients (age02=0212.002±025.102years) treated with quetiapine (dose02=0247702±0221202mg, duration02=0222.002±0210.102weeks) were identified and six were judged to be responders based on impressions from chart review and Conners Parent Scale (CPS). Improvements were observed in the conduct, inattention, and hyperactivity subscales of the CPS. Adverse events were mild with sedation being the most common, and no patient required treatment termination. Quetiapine may be beneficial in children and adolescents with pervasive developmental disorders, however open-label and double-blind, placebo-controlled studies are warranted.
The genetic landscapes of autism spectrum disorders
DOI:10.1146/annurev-genom-091212-153431
URL
PMID:23875794
[本文引用: 1]
The autism spectrum disorders (ASD) are characterized by impairments in social interaction and stereotyped behaviors. For the majority of individuals with ASD, the causes of the disorder remain unknown; however, in up to 25% of cases, a genetic cause can be identified. Chromosomal rearrangements as well as rare and de novo copy-number variants are present in similar to 10-20% of individuals with ASD, compared with 1-2% in the general population and/or unaffected siblings. Rare and de novo coding-sequence mutations affecting neuronal genes have also been identified in similar to 5-10% of individuals with ASD. Common variants such as single-nucleotide polymorphisms seem to contribute to ASD susceptibility, but, taken individually, their effects appear to be small. Despite a heterogeneous genetic landscape, the genes implicated thus far-which are involved in chromatin remodeling, metabolism, mRNA translation, and synaptic function-seem to converge in common pathways affecting neuronal and synaptic homeostasis. Animal models developed to study these genes should lead to a better understanding of the diversity of the genetic landscapes of ASD.
A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism
DOI:10.1186/2040-2392-2-1
URL
PMID:21247446
[本文引用: 1]
Background Genome-wide Association Studies (GWAS) have proved invaluable for the identification of disease susceptibility genes. However, the prioritization of candidate genes and regions for follow-up studies often proves difficult due to false-positive associations caused by statistical noise and multiple-testing. In order to address this issue, we propose the novel GWAS noise reduction (GWAS-NR) method as a way to increase the power to detect true associations in GWAS, particularly in complex diseases such as autism. Methods GWAS-NR utilizes a linear filter to identify genomic regions demonstrating correlation among association signals in multiple datasets. We used computer simulations to assess the ability of GWAS-NR to detect association against the commonly used joint analysis and Fisher's methods. Furthermore, we applied GWAS-NR to a family-based autism GWAS of 597 families and a second existing autism GWAS of 696 families from the Autism Genetic Resource Exchange (AGRE) to arrive at a compendium of autism candidate genes. These genes were manually annotated and classified by a literature review and functional grouping in order to reveal biological pathways which might contribute to autism aetiology. Results Computer simulations indicate that GWAS-NR achieves a significantly higher classification rate for true positive association signals than either the joint analysis or Fisher's methods and that it can also achieve this when there is imperfect marker overlap across datasets or when the closest disease-related polymorphism is not directly typed. In two autism datasets, GWAS-NR analysis resulted in 1535 significant linkage disequilibrium (LD) blocks overlapping 431 unique reference sequencing (RefSeq) genes. Moreover, we identified the nearest RefSeq gene to the non-gene overlapping LD blocks, producing a final candidate set of 860 genes. Functional categorization of these implicated genes indicates that a significant proportion of them cooperate in a coherent pathway that regulates the directional protrusion of axons and dendrites to their appropriate synaptic targets. Conclusions As statistical noise is likely to particularly affect studies of complex disorders, where genetic heterogeneity or interaction between genes may confound the ability to detect association, GWAS-NR offers a powerful method for prioritizing regions for follow-up studies. Applying this method to autism datasets, GWAS-NR analysis indicates that a large subset of genes involved in the outgrowth and guidance of axons and dendrites is implicated in the aetiology of autism.
Acetylcholine elevation relieves cognitive rigidity and social deficiency in a mouse model of autism
DOI:10.1038/npp.2013.274
URL
PMID:24096295
[本文引用: 3]
Abstract Autism spectrum disorders (ASD) are defined by behavioral deficits in social interaction and communication, repetitive stereotyped behaviors, and restricted interests/cognitive rigidity. Recent studies in humans and animal-models suggest that dysfunction of the cholinergic system may underlie autism-related behavioral symptoms. Here we tested the hypothesis that augmentation of acetylcholine (ACh) in the synaptic cleft by inhibiting acetylcholinesterase may ameliorate autistic phenotypes. We first administered the acetylcholinesterase inhibitor (AChEI) Donepezil systemically by intraperitoneal (i.p.) injections. Second, the drug was injected directly into the rodent homolog of the caudate nucleus, the dorsomedial striatum (DMS), of the inbred mouse strain BTBR T+tf/J (BTBR), a commonly-used model presenting all core autism-related phenotypes and expressing low brain ACh levels. We found that i.p. injection of AChEI to BTBR mice significantly relieved autism-relevant phenotypes, including decreasing cognitive rigidity, improving social preference, and enhancing social interaction, in a dose-dependent manner. Microinjection of the drug directly into the DMS, but not into the ventromedial striatum, led to significant amelioration of the cognitive-rigidity and social-deficiency phenotypes. Taken together, these findings provide evidence of the key role of the cholinergic system and the DMS in the etiology of ASD, and suggest that elevated cognitive flexibility may result in enhanced social attention. The potential therapeutic effect of AChEIs in ASD patients is discussed.
Subchronic treatment of donepezil rescues impaired social, hyperactive, and stereotypic behavior in valproic acid-induced animal model of autism
DOI:10.1371/journal.pone.0104927
URL
PMID:25133713
[本文引用: 1]
Autism spectrum disorder (ASD) is a group of pervasive developmental disorders with core symptoms such as sociability deficit, language impairment, and repetitive/restricted behaviors. Although worldwide prevalence of ASD has been increased continuously, therapeutic agents to ameliorate the core symptoms especially social deficits, are very limited. In this study, we investigated therapeutic potential of donepezil for ASD using valproic acid-induced autistic animal model (VPA animal model). We found that prenatal exposure of valproic acid (VPA) induced dysregulation of cholinergic neuronal development, most notably the up-regulation of acetylcholinesterase (AChE) in the prefrontal cortex of affected rat and mouse offspring. Similarly, differentiating cortical neural progenitor cell in culture treated with VPA showed increased expression of AChE in vitro. Chromatin precipitation experiments revealed that acetylation of histone H3 bound to AChE promoter region was increased by VPA. In addition, other histone deacetyalse inhibitors (HDACIs) such as trichostatin A and sodium butyrate also increased the expression of AChE in differentiating neural progenitor cells suggesting the essential role of HDACIs in the regulation of AChE expression. For behavioral analysis, we injected PBS or donepezil (0.3 mg/kg) intraperitoneally to control and VPA mice once daily from postnatal day 14 all throughout the experiment. Subchronic treatment of donepezil improved sociability and prevented repetitive behavior and hyperactivity of VPA-treated mice offspring. Taken together, these results provide evidence that dysregulation of ACh system represented by the up-regulation of AChE may serve as an effective pharmacological therapeutic target against autistic behaviors in VPA animal model of ASD, which should be subjected for further investigation to verify the clinical relevance.
Topoisomerases facilitate transcription of long genes linked to autism
DOI:10.1038/nature12504
URL
PMID:3767287
[本文引用: 1]
Abstract Topoisomerases are expressed throughout the developing and adult brain and are mutated in some individuals with autism spectrum disorder (ASD). However, how topoisomerases are mechanistically connected to ASD is unknown. Here we find that topotecan, a topoisomerase 1 (TOP1) inhibitor, dose-dependently reduces the expression of extremely long genes in mouse and human neurons, including nearly all genes that are longer than 200 ilobases. Expression of long genes is also reduced after knockdown of Top1 or Top2b in neurons, highlighting that both enzymes are required for full expression of long genes. By mapping RNA polymerase II density genome-wide in neurons, we found that this length-dependent effect on gene expression was due to impaired transcription elongation. Interestingly, many high-confidence ASD candidate genes are exceptionally long and were reduced in expression after TOP1 inhibition. Our findings suggest that chemicals and genetic mutations that impair topoisomerases could commonly contribute to ASD and other neurodevelopmental disorders.
What do genetic studies tell us about the heritable basis of common epilepsy? Polygenic or complex epilepsy?
DOI:10.1016/j.neulet.2017.03.042
URL
PMID:28347857
[本文引用: 1]
The search for genes associated with common epilepsy, including both focal and generalised epilepsies, has been intensive in the past few decades. Consequently, our understanding of the genetic background of common epilepsy has improved considerably, and current genetic studies have optimised their design accordingly, showing much promise for the future. Nevertheless, we can only explain a fraction of the heritability of common epilepsy with the currently known genetic factors. These factors have been identified with a range of different gene mapping techniques, including linkage analysis of epilepsy families, association studies, and recent large scale sequencing studies, which individually are optimal to detect a certain class of genetic variation. Here, we give a selected overview of the genetic studies that illustrate the evolution of epilepsy genetics and contribute to the evidence for a polygenic basis of common epilepsy that likely involves both rare and common disease variants.
Nicotinic receptor abnormalities in the cerebellar cortex in autism
DOI:10.1093/brain/awf160
URL
PMID:12076999
[本文引用: 1]
Autism is a common developmental disorder associated with structural and inferred neurochemical abnormalities of the brain. Cerebellar abnormalities frequently have been identified, based on neuroimaging or neuropathology. Recently, the cholinergic neurotransmitter system has been implicated on the basis of nicotinic receptor loss in the cerebral cortex. Cerebellar cholinergic activities were therefore investigated in autopsy tissue from a series of autistic individuals. The presynaptic cholinergic enzyme, choline acetyltransferase, together with nicotinic and muscarinic receptor subtypes were compared in the cerebellum from age-matched mentally retarded autistic (eight), normal control (10) and non-autistic mentally retarded individuals (11). The nicotinic receptor binding the agonist epibatidine (the high affinity receptor subtype, consisting primarily of alpha3 and alpha4, together with beta2 receptor subunits) was significantly reduced by 40-50% in the granule cell, Purkinje and molecular layers in the autistic compared with the normal group (P < 0.05). There was an opposite increase (3-fold) in the nicotinic receptor binding alpha-bungarotoxin (to the alpha7 subunit) which reached significance in the granule cell layer (P < 0.05). These receptor changes were paralleled by a significant reduction (P < 0.05) and non-significant increase, respectively, of alpha4 and alpha7 receptor subunit immunoreactivity measured using western blotting. Immunohistochemically loss of alpha(4 )reactivity was apparent from Purkinje and the other cell layers, with increased alpha7 reactivity in the granule cell layer. There were no significant changes in choline acetyltransferase activity, or in muscarinic M1 and M2 receptor subtypes in autism. In the non-autistic mentally retarded group, the only significant abnormality was a reduction in epibatidine binding in the granule cell and Purkinje layers. In two autistic cases examined histologically, Purkinje cell loss was observed in multiple lobules throughout the vermis and hemispheres. This was more severe in one case with epilepsy, which also showed vermis folial malformation. The case with less severe Purkinje cell loss also showed cerebellar white matter thinning and demyelination. These findings indicate a loss of the cerebellar nicotinic alpha4 receptor subunit in autism which may relate to the loss of Purkinje cells, and a compensatory increase in the alpha7 subunit. It remains to be determined how these receptor abnormalities are involved in neurodevelopment in autism and what is the relationship to mental function. Since nicotinic receptor agonists enhance attentional function and also induce an elevation in the high affinity receptor, nicotinic therapy in autism may be worth considering.
Localization of muscarinic m3 receptor protein and M3 receptor binding in rat brain
DOI:10.1016/0306-4522(94)90017-5
URL
PMID:7898649
[本文引用: 1]
The results confirm the presence of m3 protein in brain, its low abundance compared to other muscarinic receptor subtypes, and provide the first immunocytochemical map of its precise localization. The distribution of m3 suggests that it mediates a wide variety of cholinergic processes in brain, inclucling possible roles in learning and memory, motor function and behavioral state control. However, since the distribution of the molecularly-defined receptor protein is distinct from the pharmacologically-defined M3 binding site, investigations of the functions of m3 in brain must await development of more selective ligands or use of non-pharmacological approaches.
Expression of m1-m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation
A family of muscarinic ACh receptor genes are expressed in hippocampus, but little is known about the of the encoded and their regulation by cholinergic . Subtype-specific were used to localize m1-m4 in the hippocampal by immunocytochemistry and to determine the alterations in the subtypes following deafferentation. Each of the receptors is differentially localized in Ammon's horn and dentate gyrus, with highly complementary distributions. m1 is widely expressed in somata and of pyramidal neurons and granule in dentate gyrus. m2 immunoreactivity is expressed mostly in nonpyramidal neurons, and in several discrete bands of fibers and puncta surrounding pyramidal neurons and other layers. m3 is enriched in pyramidal neurons, the neuropil in stratum lacunosum-moleculare and the outer third of the molecular layer of dentate gyrus. m4 is enriched in nonpyramidal neurons, in fiber pathways (alveus, , and hippocampal commissure), and in the inner third of the molecular layer. -fornix lesions decreased ipsilateral m2- and m4-immunoreactive in the , with no apparent changes in the distribution of any of the receptors in hippocampus. 192-IgG immunotoxin lesions of the cholinergic septohippocampal projections, which spare noncholinergic projections, produced a small decrease in m2-immunoreactive fibers in the with no other major changes in the distribution of subtypes. Immunoprecipitation studies at 3-28 d following -fornix lesions revealed a 25% loss of m2 at 3 d in hippocampus, and upregulation of both m1 (20-29% at 7-14 d) and m4 (44% at 28 d). Thus, the vast majority of muscarinic receptor subtypes are intrinsic to the hippocampal and/or nonseptal hippocampal afferents. A subset of m2 and m4 are presynaptically localized, with m2 in cholinergic and m2 and m4 possibly in noncholinergic that comprise the septohippocampal pathway. The unique laminar and regional distributions of m1-m4 in the hippocampus reflect differential cellular and subcellular distributions of the subtypes and/or selective association of receptor subtypes with certain afferent and intrinsic connections. These results indicate that each subtype likely has a different role in cholinergic modulation of excitatory and inhibitory hippocampal circuits.
Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders
DOI:10.1038/mp.2017.140
URL
PMID:28831199
[本文引用: 2]
Abstract Autism spectrum disorder (ASD) represents a set of complex neurodevelopmental disorders with large degrees of heritability and heterogeneity. We sequenced 136 microcephaly or macrocephaly (Mic-Mac)-related genes and 158 possible ASD-risk genes in 536 Chinese ASD probands and detected 22 damaging de novo mutations (DNMs) in 20 genes, including CHD8 and SCN2A, with recurrent events. Nine of the 20 genes were previously reported to harbor DNMs in ASD patients from other populations, while 11 of them were first identified in present study. We combined genetic variations of the 294 sequenced genes from publicly available whole-exome or whole-genome sequencing studies (4167 probands plus 1786 controls) with our Chinese population (536 cases plus 1457 controls) to optimize the power of candidate-gene prioritization. As a result, we prioritized 67 ASD-candidate genes that exhibited significantly higher probabilities of haploinsufficiency and genic intolerance, and significantly interacted and co-expressed with each another, as well as other known ASD-risk genes. Probands with DNMs or rare inherited mutations in the 67 candidate genes exhibited significantly lower intelligence quotients, supporting their strong functional impact. In addition, we prioritized 39 ASD-related Mic-Mac-risk genes, and showed their interaction and co-expression in a functional network that converged on chromatin remodeling, synapse transmission and cell cycle progression. Genes within the three functional subnetworks exhibited distinct and recognizable spatiotemporal-expression patterns in human brains and laminar-expression profiles in the developing neocortex, highlighting their important roles in brain development. Our results indicate some of Mic-Mac-risk genes are involved in ASD.
Function and regulation of CREB family transcription factors in the nervous system
Breakpoint mapping and haplotype analysis of translocation t(1;12) (q43;q21.1) in two apparently independent families with vascular phenotypes
DOI:10.1002/mgg3.346
URL
PMID:29168350
[本文引用: 2]
Abstract Background The risk of serious congenital anomaly for de novo balanced translocations is estimated to be at least 6%. We identified two apparently independent families with a balanced t(1;12)(q43;q21.1) as an outcome of a “Systematic Survey of Balanced Chromosomal Rearrangements in Finns.” In the first family, carriers ( n =6) manifest with learning problems in childhood, and later with unexplained neurological symptoms (chronic headache, balance problems, tremor, fatigue) and cerebral infarctions in their 50s. In the second family, two carriers suffer from tetralogy of Fallot, one from transient ischemic attack and one from migraine. The translocation cosegregates with these vascular phenotypes and neurological symptoms. Methods and Results We narrowed down the breakpoint regions using mate pair sequencing. We observed conserved haplotypes around the breakpoints, pointing out that this translocation has arisen only once. The chromosome 1 breakpoint truncates a CHRM3 processed transcript, and is flanked by the 5′ end of CHRM3 and the 3′ end of RYR2 . TRHDE , KCNC2 , and ATXN7L3B flank the chromosome 12 breakpoint. Conclusions This study demonstrates a balanced t(1;12)(q43;q21.1) with conserved haplotypes on the derived chromosomes. The translocation seems to result in vascular phenotype, with or without neurological symptoms, in at least two families. We suggest that the translocation influences the positional expression of CHRM3 , RYR2 , TRHDE , KCNC2 , and/or ATXN7L3B .
Abnormal mGlu 5 receptor/endocannabinoid coupling in mice lacking FMRP and BC1 RNA
DOI:10.1038/npp.2010.19
URL
PMID:20393458
[本文引用: 1]
Transcriptional silencing of the gene encoding the fragile X mental retardation protein (FMRP) causes fragile X syndrome (FXS). FMRP acts as a translational repressor at central synapses, and molecular and synaptic plasticity studies have shown that the absence of this protein alters metabotropic glutamate 5 receptors (mGlu5Rs)-mediated signaling. In the striatum of mice lacking FMRP, we found enhanced activity of diacylglycerol lipase (DAGL), the enzyme limiting 2-arachidonoylglicerol (2-AG) synthesis, associated with altered sensitivity of GABA synapses to the mobilization of this endocannabinoid by mGlu5R stimulation with DHPG. Mice lacking another repressor of synaptic protein synthesis, BC1 RNA, also showed potentiated mGlu5R-driven 2-AG responses, indicating that both FMRP and BC1 RNA act as physiological constraints of mGlu5R/endocannabinoid coupling at central synapses. The effects of FMRP ablation on DAGL activity and on DHPG-mediated inhibition of GABA synapses were enhanced by simultaneous genetic inactivation of FMRP and BC1 RNA. In double FMRP and BC1 RNA lacking mice, striatal levels of 2-AG were also enhanced compared with control animals and to single mutants. Our data indicate for the first time that mGlu5R-driven endocannabinoid signaling in the striatum is under the control of both FMRP and BC1 RNA. The abnormal mGlu5R/2-AG coupling found in FMRP-KO mice emphasizes the involvement of mGlu5Rs in the synaptic defects of FXS, and identifies the modulation of the endocannabinoid system as a novel target for the treatment of this severe neuropsychiatric disorder.
Open-label quetiapine in the treatment of children and adolescents with autistic disorder
Molecular analysis of nicotinic receptor expression in autism
DOI:10.1016/j.molbrainres.2004.01.003
URL
PMID:15046869
[本文引用: 1]
Autism is a developmental disorder of unknown aetiopathology and lacking any specific pharmacological therapeutic intervention. Neurotransmitters such as serotonin, gamma-aminobutyric acid (GABA) and acetylcholine have been implicated. Abnormalities in nicotinic acetylcholine receptors have been identified including cortical loss of binding to the α4/β2 subtype and increase in cerebellar α7 binding. Receptor expression (mRNA) has not so far been systematically examined. This study aims to further explore the role of nicotinic receptors in autism by analysing nicotinic receptor subunit mRNA in conjunction with protein levels and receptor binding in different brain areas. Quantitative RT-PCR for α4, α7 and β2 subunit mRNA expression levels; α3, α4, α7 and β2 subunit protein expression immunochemistry and specific radioligand receptor binding were performed in adult autism and control brain samples from cerebral cortex and cerebellum. Alpha4 and β2 protein expression and receptor binding density as well as α4 mRNA levels were lower in parietal cortex in autism, while α7 did not change for any of these parameters. In cerebellum, α4 mRNA expression was increased, whereas subunit protein and receptor levels were decreased. Alpha7 receptor binding in cerebellum was increased alongside non-significant elevations in mRNA and protein expression levels. No significant changes were found for β2 in cerebellum. The data obtained, using complementary measures of receptor expression, indicate that reduced gene expression of the α4β2 nicotinic receptor in the cerebral cortex is a major feature of the neurochemical pathology of autism, whilst post-transcriptional abnormalities of both this and the α7 subtype are apparent in the cerebellum. The findings point to dendritic and/or synaptic nicotinic receptor abnormalities that may relate to disruptions in cerebral circuitry development.
Mapping of five subtype genes for muscarinic acetylcholine receptor to mouse chromosomes
DOI:10.1266/ggs.74.15
URL
PMID:10549128
[本文引用: 1]
Muscarinic acetylcholine receptors in mammals consist of five subtypes (M1-M5) encoded by distinct genes. They are widely expressed throughout the body and play a variety of roles in the peripheral and central nervous systems. Although their pharmacological properties have been studied extensively in vitro, colocalization of the multiple subtypes in each tissue and lack of subtype-specific ligands have hampered characterization of the respective subtypes in vivo. We have mapped mouse genomic loci for all five genes (Chrm1-5) by restriction fragment length variant (RFLV) analyses in interspecific backcross mice. Chrm1, Chrm2, and Chrm3 were mapped to chromosome (Chr) 19, 6, and 13, respectively. Both Chrm4 and Chrm5 were mapped to Chr 2. Although a comparison of their map positions with other mutations in their vicinities suggested a possibility that the El2 (epilepsy 2) allele might be a mutation in Chrm5, sequencing analyses of the Chrm5 gene in the El2 mutant mice did not support such a hypothesis.
Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype
DOI:10.1073/pnas.97.17.9579
URL
[本文引用: 2]
Muscarinic acetylcholine receptors consist of five distinct subtypes and have been important targets for drug development. In the periphery, muscarinic acetylcholine receptors mediate cholinergic signals to autonomic organs, but specific physiological functions of each subtype remain poorly elucidated. Here, we have constructed and analyzed mutant mice lacking the M3receptor and have demonstrated that this subtype plays key roles in salivary secretion, pupillary constriction, and bladder detrusor contractions. However, M3-mediated signals in digestive and reproductive organs are dispensable, likely because of redundant mechanisms through other muscarinic acetylcholine receptor subtypes or other mediators. In addition, we have found prominent urinary retention only in the male, which indicates a considerable sex difference in the micturition mechanism. Accordingly, this mutant mouse should provide a useful animal model for investigation of human diseases that are affected in the peripheral cholinergic functions.
The BTBR mouse model of autism spectrum disorders has learning and attentional impairments and alterations in acetylcholine and kynurenic acid in prefrontal cortex
DOI:10.1371/journal.pone.0062189
URL
PMID:3634761
[本文引用: 1]
Autism is a complex spectrum of disorders characterized by core behavioral deficits in social interaction, communication, repetitive stereotyped behaviors and restricted interests. Autism frequently presents with additional cognitive symptoms, including attentional deficits and intellectual disability. Preclinical models are important tools for studying the behavioral domains and biological underpinnings of autism, and potential treatment targets. The inbred BTBR T+tf/J (BTBR) mouse strain has been used as an animal model of core behavioral deficits in autism. BTBR mice exhibit repetitive behaviors and deficits in sociability and communication, but other aspects of their cognitive phenotype, including attentional performance, are not well characterized. We examined the attentional abilities of BTBR mice in the 5-choice serial reaction time task (5-CSRTT) using an automated touchscreen testing apparatus. The 5-CSRTT is an analogue of the human continuous performance task of attention, and so both the task and apparatus have translational relevance to human touchscreen cognitive testing. We also measured basal extracellular levels of a panel of neurotransmitters within the medial prefrontal cortex, a brain region critically important for performing the 5-CSRTT. We found that BTBR mice have increased impulsivity, defined as an inability to withhold responding, and decreased motivation, as compared to C57Bl/6J mice. Both of these features characterize attentional deficit disorders in humans. BTBR mice also display decreased accuracy in detecting short stimuli, lower basal levels of extracellular acetylcholine and higher levels of kynurenic acid within the prefrontal cortex. Intact cholinergic transmission in prefrontal cortex is required for accurate performance of the 5-CSRTT, consequently this cholinergic deficit may underlie less accurate performance in BTBR mice. Based on our findings that BTBR mice have attentional impairments and alterations in a key neural substrate of attention, we propose that they may be valuable for studying mechanisms for treatment of cognitive dysfunction in individuals with attention deficits and autism.
Growth restriction, leptin, and the programming of adult behavior in mice
DOI:10.1016/j.bbr.2014.08.054
URL
PMID:4252372
[本文引用: 2]
Prematurity and neonatal growth restriction (GR) are risk factors for autism and attention deficit hyperactivity disorder (ADHD). Leptin production is suppressed during periods of undernutrition, and we have shown that isolated neonatal leptin deficiency leads to adult hyperactivity while neonatal leptin supplementation normalizes the brain morphology of GR mice. We hypothesized that neonatal leptin would prevent the development of GR-associated behavioral abnormalities. From postnatal day 4–14, C57BL/6 mice were randomized to daily injections of saline or leptin (80ng/g), and GR was identified by a weanling weight below the tenth percentile. The behavioral phenotypes of GR and control mice were assessed beginning at 4 months. Within the tripartite chamber, GR mice had significantly impaired social interaction. Baseline escape times from the Barnes maze were faster for GR mice (65+/616s vs 87+/617s for controls, p<0.05), but GR mice exhibited regression in their escape times on days 2 and 3 (56% regressed vs 22% of control saline mice, p<0.05). Compared to controls, GR mice entered the open arms of the elevated plus maze more often and stayed there longer (72+/6110s vs 36+/615s, p<0.01). Neonatal leptin supplementation normalized the behavior of GR mice across all behavioral assays. In conclusion, GR alters the social interactions, learning and activity of mice, and supplementation with the neurotrophic hormone leptin mitigates these effects. We speculate neonatal leptin deficiency may contribute to the adverse neurodevelopmental outcomes associated with postnatal growth restriction, and postnatal leptin therapy may be protective.
Whole-genome sequencing in autism identifies hot spots for de novo germline mutation
GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism
DOI:10.1371/journal.pone.0009706
URL
PMID:2838793
[本文引用: 1]
Nearly 1% of children in the United States exhibit autism spectrum disorders, but causes and treatments remain to be identified. Mice with deletion of thefragile X mental retardation 1(Fmr1) gene are used to model autism because loss ofFmr1gene function causes Fragile X Syndrome (FXS) and many people with FXS exhibit autistic-like behaviors. Glycogen synthase kinase-3 (GSK3) is hyperactive in brains ofFmr1knockout mice, and inhibition of GSK3 by lithium administration ameliorates some behavioral impairment in these mice. We extended our studies of this association by testing whether GSK3 contributes to socialization behaviors. This used two mouse models with disrupted regulation of GSK3,Fmr1knockout mice and GSK3 knockin mice, in which inhibitory serines of the two isoforms of GSK3, GSK3 and GSK3 , are mutated to alanines, leaving GSK3 fully active. To assess sociability, test mice were introduced to a restrained stimulus mouse (S1) for 10 min, followed by introduction of a second restrained stimulus mouse (S2) for 10 min, which assesses social preference.Fmr1knockout and GSK3 knockin mice displayed no deficit in sociability with the S1 mouse, but unlike wild-type mice neither demonstrated social preference for the novel S2 mouse.Fmr1knockout mice displayed more anxiety-related behaviors during social interaction (grooming, rearing, and digging) than wild-type mice, which was ameliorated by inhibition of GSK3 with chronic lithium treatment. These results indicate that impaired inhibitory regulation of GSK3 inFmr1knockout mice may contribute to some socialization deficits and that lithium treatment can ameliorate certain socialization impairments. As discussed in the present work, these results suggest a role for GSK3 in social behaviors and implicate inhibition of GSK3 as a potential therapeutic.
Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome
DOI:10.1523/JNEUROSCI.2623-05.2006
URL
PMID:16399702
[本文引用: 1]
Abstract Loss-of-function mutations or abnormal expression of the X-linked gene encoding methyl CpG binding protein 2 (MeCP2) cause a spectrum of postnatal neurodevelopmental disorders including Rett syndrome (RTT), nonsyndromic mental retardation, learning disability, and autism. Mice expressing a truncated allele of Mecp2 (Mecp2(308)) reproduce the motor and social behavior abnormalities of RTT; however, it is not known whether learning deficits are present in these animals. We investigated learning and memory, neuronal morphology, and synaptic function in Mecp2(308) mice. Hippocampus-dependent spatial memory, contextual fear memory, and social memory were significantly impaired in Mecp2(308) mutant males (Mecp2(308/Y)). The morphology of dendritic arborizations, the biochemical composition of synaptosomes and postsynaptic densities, and brain-derived neurotrophic factor expression were not altered in these mice. However, reduced postsynaptic density cross-sectional length was identified in asymmetric synapses of area CA1 of the hippocampus. In the hippocampus of symptomatic Mecp2(308/Y) mice, Schaffer-collateral synapses exhibited enhanced basal synaptic transmission and decreased paired-pulse facilitation, suggesting that neurotransmitter release was enhanced. Schaffer-collateral long-term potentiation (LTP) was impaired. LTP was also reduced in the motor and sensory regions of the neocortex. Finally, very early symptomatic Mecp2(308/Y) mice had increased basal synaptic transmission and deficits in the induction of long-term depression. These data demonstrate a requirement for MeCP2 in learning and memory and suggest that functional and ultrastructural synaptic dysfunction is an early event in the pathogenesis of RTT.
Patterns and rates of exonic de novo mutations in autism spectrum disorders
Neuropeptides and neurotrophins in neonatal blood of children with autism or mental retardation
DOI:10.1002/ana.1024
URL
PMID:11357950
[本文引用: 1]
There has been little exploration of major biologic regulators of cerebral development in autism. In archived neonatal blood of children with autistic spectrum disorders (n = 69), mental retardation without autism (n = 60), or cerebral palsy (CP, n = 63) and of control children (n = 54), we used recycling immunoaffinity chromatography to measure the neuropeptides substance P (SP), vasoactive intestinal peptide (VIP), pituitary adenylate cyclase-activating polypeptide (PACAP), calcitonin gene-related peptide (CGRP), and the neurotrophins nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT3), and neurotrophin 4/5 (NT4/5). Neonatal concentrations of VIP, CGRP, BDNF, and NT4/5 were higher (ANOVA, all p values < 0.0001 by Scheffe test for pairwise differences) in children in the autistic spectrum and in those with mental retardation without autism than in control children. In 99% of children with autism and 97% with mental retardation, levels of at least one of these substances exceeded those of all control children. Concentrations were similar in subgroups of the autistic spectrum (core syndrome with or without mental retardation, other autistic spectrum disorders with or without mental retardation) and in the presence or absence of a history of regression. Among children with mental retardation, concentrations did not differ by severity or known cause (n = 11, including 4 with Down syndrome). Concentrations of measured substances were similar in children with CP as compared with control subjects. SP, PACAP, NGF, and NT3 were not different by diagnostic group. No measured analyte distinguished children with autism from children with mental retardation alone. In autism and in a heterogeneous group of disorders of cognitive function, overexpression of certain neuropeptides and neurotrophins was observed in peripheral blood drawn in the first days of life.
Synaptic basis of social dysfunction: A focus on postsynaptic proteins linking group-I mGluRs with AMPARs and NMDARs
DOI:10.1111/ejn.12510
URL
PMID:24712991
[本文引用: 1]
Most of us engage in social interactions on a daily basis and the repertoire of social behaviors we acquire during development and later in life are incredibly varied. However, in many neurodevelopmental disorders, including autism spectrum disorders (ASDs), social behavior is severely compromised and indeed this represents a key diagnostic component for such conditions. From genetic association studies, it is increasingly apparent that genes identified as altered in individuals with ASDs often encode synaptic proteins. Moreover, these synaptic proteins typically serve to scaffold group-I metabotropic glutamate receptors (group-I mGluRs) and ionotropic glutamate receptors (iGluRs; AMPARs and NMDARs), or to enable group-I mGluR to iGluR crosstalk via protein synthesis. Here we aim to explore the possibility of a causal link between altered function of such synaptic proteins and impaired social behaviors that feature in neurodevelopmental disorders, such as ASDs. We review the known synaptic function and role in social behaviors of selected post-synaptic structural proteins (Shank, SAPAP and neuroligin) and regulators of protein synthesis (TSC1/2, FMRP and PTEN). While manipulations of proteins involved in group-I mGluR and iGluR scaffolding or crosstalk frequently lead to profound alterations in synaptic function and one or more components of social behavior, the neuronal circuits responsible for impairments in specific social behaviors are often poorly defined. We argue for an improved understanding of the neuronal circuits underlying specific social behaviors to aid the development of new ASD therapies.
Synaptic Wnt signaling-a contributor to major psychiatric disorders?
DOI:10.1007/s11689-011-9083-6
URL
PMID:3180925
[本文引用: 1]
Wnt signaling is a key pathway that helps organize development of the nervous system. It influences cell proliferation, cell fate, and cell migration in the developing nervous system, as well as axon guidance, dendrite development, and synapse formation. Given this wide range of roles, dysregulation of Wnt signaling could have any number of deleterious effects on neural development and thereby contribute in many different ways to the pathogenesis of neurodevelopmental disorders. Some major psychiatric disorders, including schizophrenia, bipolar disorder, and autism spectrum disorders, are coming to be understood as subtle dysregulations of nervous system development, particularly of synapse formation and maintenance. This review will therefore touch on the importance of Wnt signaling to neurodevelopment generally, while focusing on accumulating evidence for a synaptic role of Wnt signaling. These observations will be discussed in the context of current understanding of the neurodevelopmental bases of major psychiatric diseases, spotlighting schizophrenia, bipolar disorder, and autism spectrum disorder. In short, this review will focus on the potential role of synapse formation and maintenance in major psychiatric disorders and summarize evidence that defective Wnt signaling could contribute to their pathogenesis via effects on these late neural differentiation processes.
Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders
DOI:10.1126/science.1227764
URL
PMID:23160955
[本文引用: 2]
Exome sequencing studies of autism spectrum disorders (ASDs) have identified many de novo mutations but few recurrently disrupted genes. We therefore developed a modified molecular inversion probe method enabling ultra-low-cost candidate gene resequencing in very large cohorts. To demonstrate the power of this approach, we captured and sequenced 44 candidate genes in 2446 ASD probands. We discovered 27 de novo events in 16 genes, 59% of which are predicted to truncate proteins or disrupt splicing. We estimate that recurrent disruptive mutations in six genes— CHD8 , DYRK1A , GRIN2B , TBR1 , PTEN , and TBL1XR1 —may contribute to 1% of sporadic ASDs. Our data support associations between specific genes and reciprocal subphenotypes ( CHD8 -macrocephaly and DYRK1A -microcephaly) and replicate the importance of a β-catenin–chromatin-remodeling network to ASD etiology.
Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations
DOI:10.1038/nature10989
URL
PMID:22495309
[本文引用: 1]
Abstract It is well established that autism spectrum disorders (ASD) have a strong genetic component; however, for at least 70% of cases, the underlying genetic cause is unknown. Under the hypothesis that de novo mutations underlie a substantial fraction of the risk for developing ASD in families with no previous history of ASD or related phenotypes--so-called sporadic or simplex families--we sequenced all coding regions of the genome (the exome) for parent-child trios exhibiting sporadic ASD, including 189 new trios and 20 that were previously reported. Additionally, we also sequenced the exomes of 50 unaffected siblings corresponding to these new (n = 31) and previously reported trios (n = 19), for a total of 677 individual exomes from 209 families. Here we show that de novo point mutations are overwhelmingly paternal in origin (4:1 bias) and positively correlated with paternal age, consistent with the modest increased risk for children of older fathers to develop ASD. Moreover, 39% (49 of 126) of the most severe or disruptive de novo mutations map to a highly interconnected -catenin/chromatin remodelling protein network ranked significantly for autism candidate genes. In proband exomes, recurrent protein-altering mutations were observed in two genes: CHD8 and NTNG1. Mutation screening of six candidate genes in 1,703 ASD probands identified additional de novo, protein-altering mutations in GRIN2B, LAMC3 and SCN1A. Combined with copy number variant (CNV) data, these results indicate extreme locus heterogeneity but also provide a target for future discovery, diagnostics and therapeutics.
Recurrence risk for autism spectrum disorders: A baby siblings research consortium study
De novo 911 Kb interstitial deletion on chromosome 1q43 in a boy with mental retardation and short stature
DOI:10.1016/j.ejmg.2011.11.004
URL
PMID:22186213
[本文引用: 2]
Patients with distal deletions of chromosome 1q have a recognizable syndrome that includes microcephaly, hypoplasia or agenesis of the corpus callosum, and psychomotor retardation. Although these symptoms have been attributed to deletions of 1q42–1q44, the minimal chromosomal region involved has not yet defined. In this report, we describe a 7 years old male with mental retardation, cryptorchid testes, short stature and alopecia carrying only an interstitial de novo deletion of 91102Kb in the 1q43 region (239,597,095–240,508,817) encompassing three genes CHRM3, RPS7P5 and FMN2.
Cholinergic activity in autism: Abnormalities in the cerebral cortex and basal forebrain
DOI:10.1176/appi.ajp.158.7.1058
URL
PMID:11431227
[本文引用: 1]
OBJECTIVE: Measures of cholinergic transmitter activity were investigated in patients with autism because of reported neuropathological abnormalities in cholinergic nuclei in the basal forebrain. METHOD: Levels of cholinergic enzyme and receptor activity were measured in the frontal and parietal cerebral cortex of deceased autistic adults, similarly aged normal adults without mental retardation, and nonautistic mentally retarded adults. The immunoreactivity levels of brain-derived neurotrophic factor and nerve growth factor were measured in the basal forebrain. RESULTS: There were no differences between the autistic and comparison groups in choline acetyltransferase or acetylcholinesterase activity in the cerebral cortex and basal forebrain or in muscarinic M(2) receptor or alpha-bungarotoxin binding within the cortex. Cortical M(1) receptor binding was up to 30% lower than normal in the autistic subjects, and the difference reached significance in the parietal cortex. In both the parietal and frontal cortices, differences in nicotinic receptors assessed by [(3)H]epibatidine binding were significant and extensive (65%-73% lower in the autistic group than in the normal subjects); there were no differences in nicotine binding in the basal forebrain. Immunochemical analysis indicated lower levels of both the alpha(4) and beta(2) nicotinic receptor subunits in the parietal cortex. The M(1) receptor abnormality was not evident in the nonautistic group with mental retardation, although the lower [(3)H]epibatidine binding was apparent. In the basal forebrain, the level of brain-derived neurotrophic factor in the autistic group was three times as high as the level of the normal group. CONCLUSIONS: These neurochemical abnormalities implicate the cholinergic system in developmental disorders such as autism and suggest the potential for intervention based on cholinergic receptor modulation.
Deletion 1q43 encompassing only CHRM3 in a patient with autistic disorder
DOI:10.1016/j.ejmg.2012.11.003
URL
PMID:23253743
[本文引用: 4]
Deletions on the distal portion of the long arm of chromosome 1 result in complex and highly variable clinical phenotypes which include intellectual disability, autism, seizures, microcephaly/craniofacial dysmorphology, corpus callosal agenesis/hypogenesis, cardiac and genital anomalies, hand and foot abnormalities and short stature. Genotype-phenotype correlation reported a minimum region of 202Mb at 1q43-q44. We report on a 3 05 year old male patient diagnosed with autistic disorder who has social withdrawal, eating problems, repetitive stereotypic behaviors including self-injurious head banging and hair pulling, and no seizures, anxiety, or mood swings. Array comparative genomic hybridization (aCGH) showed an interstitial deletion of 47302kb at 1q43 region (239,412,391–239,885,394; NCBI build37/hg19) harboring only CHRM3 (Acetylcholine Receptor, Muscarinic, 3; OMIM: 118494). Recently, another case with a de novo interstitial deletion of 91102kb at 1q43 encompassing three genes including CHRM3 was reported. The M3 muscarinic receptor influences a multitude of central and peripheral nervous system processes via its interaction with acetylcholine and may be an important modulator of behavior, learning and memory. We propose CHRM3 as a candidate gene responsible for our patient's specific phenotype as well as the overlapping phenotypic features of other patients with 1q43 or 1q43-q44 deletions.
Convergence of genes and cellular pathways dysregulated in autism spectrum disorders
Functional impact of global rare copy number variation in autism spectrum disorders
The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/arrestin-dependent manner
DOI:10.1073/pnas.0914801107
URL
PMID:20439723
[本文引用: 1]
Degeneration of the cholinergic system is considered to be the underlying pathology that results in the cognitive deficit in Alzheimer's disease. This pathology is thought to be linked to a loss of signaling through the cholinergic M69-muscarinic receptor subtype. However, recent studies have cast doubt on whether this is the primary receptor mediating cholinergic-hippocampal learning and memory. The current study offers an alternative mechanism involving the M61-muscarinic receptor that is expressed in numerous brain regions including the hippocampus. We demonstrate here that M61-muscarinic receptor knockout mice show a deficit in fear conditioning learning and memory. The mechanism used by the M61-muscarinic receptor in this process involves receptor phosphorylation because a knockin mouse strain expressing a phosphorylation-deficient receptor mutant also shows a deficit in fear conditioning. Consistent with a role for receptor phosphorylation, we demonstrate that the M61-muscarinic receptor is phosphorylated in the hippocampus following agonist treatment and following fear conditioning training. Importantly, the phosphorylation-deficient M61-muscarinic receptor was coupled normally to G q/11 -signaling but was uncoupled from phosphorylation-dependent processes such as receptor internalization and arrestin recruitment. It can, therefore, be concluded that M61-muscarinic receptor—dependent learning and memory depends, at least in part, on receptor phosphorylation/arrestin signaling. This study opens the potential for biased M61-muscarinic receptor ligands that direct phosphorylation/arrestin-dependent (non-G protein) signaling as being beneficial in cognitive disorders.
A role for melanin-concentrating hormone in the central regulation of feeding behaviour
Neuronal nicotinic acetylcholine receptor subunits in autism: An immunohistochemical investigation in the thalamus
DOI:10.1016/j.nbd.2005.01.017
URL
PMID:16023579
[本文引用: 1]
The cholinergic system has been implicated in the development of autism on the basis of neuronal nicotinic acetylcholine receptor (nAChR) losses in cerebral and cerebellar cortex. In the present study, the first to explore nAChRs in the thalamus in autism, α4, α7 and β2 nAChR subunit expression in thalamic nuclei of adult individuals with autism ( n = 3) and age-matched control cases ( n = 3) was investigated using immunochemical methods. Loss of α7- and β2- (but not α4-) immunoreactive neurons occurred in the paraventricular nucleus (PV) and nucleus reuniens in autism. Preliminary results indicated glutamic acid decarboxylase immunoreactivity occurred at a low level in PV, co-expressed with α7 in normal and autistic cases and was not reduced in autism. This suggested loss of neuronal α7 in autism is not caused by loss of GABAergic neurons. These findings indicate nicotinic abnormalities that occur in the thalamus in autism which may contribute to sensory or attentional deficits.
Levels of cerebrospinal fluid nerve-growth factor differ in infantile autism and Rett syndrome
DOI:10.1111/j.1469-8749.1999.tb00573.x
URL
PMID:10210246
[本文引用: 1]
Autism and Rett syndrome (RS) are both developmental disorders of unknown origin. Autism is a behaviourally defined syndrome. RS, which affects girls only, is characterized by a profound learning disability following early normal development, with a consistent cluster of clinical features. Differentiation of RS from infantile autism in the very early stages of the disorders is not always easy. Both syndromes still lack discriminative laboratory markers for accurate diagnosis and differentiation. We decided to compare the CSF nerve-growth factor (NGF) levels of children with infantile autism and children with RS using enzyme-linked immunosorbent assay (ELISA). Our findings of mainly normal CSF NGF in autism and low to negligible values in RS are in agreement with the different morphological and neurochemical findings (brain growth, affected brain areas, neurotransmitter metabolism) in the two syndromes. CSF NGF could be used as a biochemical marker for differentiation of patients with autism from those with RS.
Muscarinic acetylcholine receptor activation blocks long-term potentiation at cerebellar parallel fiber-Purkinje cell synapses via cannabinoid signaling
DOI:10.1073/pnas.1221803110
URL
[本文引用: 1]
Muscarinic acetylcholine receptors (mAChRs) are known to modulate synaptic plasticity in various brain areas. A signaling pathway triggered by mAChR activation is the production and release of endocannabinoids that bind to type 1 cannabinoid receptors (CB1R) located on synaptic terminals. Using whole-cell patch-clamp recordings from rat cerebellar slices, we have demonstrated that the muscarinic agonist oxotremorine-m (oxo-m) blocks the induction of presynaptic long-term potentiation (LTP) at parallel fiber (PF)-Purkinje cell synapses in a CB1R-dependent manner. Under control conditions, LTP was induced by delivering 120 PF stimuli at 8 Hz. In contrast, no LTP was observed when oxo-m was present during tetanization. PF-LTP was restored when the CB1R antagonist N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1- piperidinyl-1H-pyrazole-3-carboxamide (AM251) was coapplied with oxo-m. Furthermore, the suppressive effect of oxo-m on PF-LTP was abrogated by the GDP analog GDP-beta-S (applied intracellularly), the phospholipase C inhibitor U-73122, and the diacylglycerol lipase inhibitor tetrahydrolipstatin (THL), suggesting that cannabinoid synthesis results from the activation of G(q)-coupled mAChRs present on Purkinje cells. The oxo-m-mediated suppression of LTP was also prevented in the presence of the M3 receptor antagonist DAU 5884, and was absent in M1/M3 receptor double-KO mice, identifying M3 receptors as primary oxo-m targets. Our findings allow for the possibility that cholinergic signaling in the cerebellum-which may result from long-term depression (LTD)-related disinhibition of cholinergic neurons in the vestibular nuclei-suppresses presynaptic LTP to prevent an up-regulation of transmitter release that opposes the reduction of postsynaptic responsiveness. This modulatory capacity of mAChR signaling could promote the functional penetrance of LTD.
The genetic relationship between individual differences in social and nonsocial behaviours characteristic of autism
DOI:10.1111/j.1467-7687.2005.00433.x
URL
PMID:16048517
[本文引用: 1]
Two types of behaviours shown in children - those reflecting social impairment and nonsocial obsessive repetitive behaviours - are central to defining and diagnosing autism spectrum disorders (ASDs). Parent and teacher data on social and nonsocial behaviours were obtained from a community sample of >3000 7-year-old twin pairs. Social and nonsocial behaviours were only modestly correlated, and it was found that some individuals had extreme scores on either social or nonsocial scales but not both. Genetic model-fitting showed that social and nonsocial behaviours are both highly heritable, but their genetic overlap is modest, with most of the genetic influence being specific to either social or nonsocial behaviours. Considering these behaviours separately might help clarify gene-brain-behaviour pathways in future research.
A de novo apparently balanced translocation [46,XY,t(2;9) (p13;p24)] interrupting RAB11FIP5 identifies a potential candidate gene for autism spectrum disorder. American Journal of Medical
DOI:10.1002/ajmg.b.30755
URL
PMID:18384058
[本文引用: 1]
Abstract Autism spectrum disorder (ASD) is a severe developmental disorder of the central nervous system characterized by impairments in social interaction, communication, and range of interests and behaviors. The syndrome's prevalence is estimated to be as high as 1 in 150 American children yet its etiology remains largely unknown. Examination of observed cytogenetic variants in individuals with ASD may identify genes involved in its pathogenesis. As part of a multidisciplinary study, an apparently balanced de novo translocation between chromosomes 2 and 9 [46,XY,t(2;9)(p13;p24)] was identified in a subject with pervasive developmental disorder not otherwise specified (PDD-NOS), and no distinctive dysmorphic features. Molecular characterization of the rearrangement revealed direct interruption of the RAB11 family interacting protein 5 (RAB11FIP5) gene. RAB11FIP5 is a Rab effector involved in protein trafficking from apical recycling endosomes to the apical plasma membrane. It is ubiquitously expressed and reported to contribute to both neurotransmitter release and neurotransmitter uptake at the synaptic junction. Detailed analysis of the rearrangement breakpoints suggests that the reciprocal translocation may have formed secondary to incorrect repair of double strand breaks (DSBs) by nonhomologous end-joining (NHEJ). (c) 2008 Wiley-Liss, Inc.
Characteristics and concordance of autism spectrum disorders among 277 twin pairs
DOI:10.1001/archpediatrics.2009.98
URL
PMID:19805709
[本文引用: 1]
To examine patterns of autism spectrum disorder (ASD) inheritance and other features in twin pairs by zygosity, sex, and specific ASD diagnosis. Cross-sectional study. Internet-based autism registry for US residents. Survey results from 277 twin pairs (210 dizygotic [DZ] and 67 monozygotic [MZ]) aged 18 years or younger with at least 1 affected twin. Zygosity and sex. Concordance within twin pairs of diagnosis, natural history, and results from standardized autism screening. Pairwise ASD concordance was 31% for DZ and 88% for MZ twins. Female and male MZ twins were 100% and 86% concordant, respectively, and DZ twin pairs with at least 1 female were less likely to be concordant (20%) than were male-male DZ twin pairs (40%). The hazard ratio for ASD diagnosis of the second twin after a first-twin diagnosis was 7.48 for MZ vs DZ twins (95% confidence interval, 3.8-14.7). Affected DZ individual twins had an earlier age at first parental concern and more frequent diagnoses of intellectual disability than did MZ twins; MZ twins had a higher prevalence of bipolar disorder and Asperger syndrome and higher concordance of the latter. Results of autism screening correlated with parent-reported ASD status in more than 90% of cases. Our data support greater ASD concordance in MZ vs DZ twins. Overall higher functioning, psychiatric comorbidity, and Asperger syndrome concordance among affected MZ vs DZ twins may also suggest differential heritability for different ASDs. For families in which one MZ twin is diagnosed with ASD, the second twin is unlikely to receive an ASD diagnosis after 12 months. In addition, Internet parent report of ASD status is valid.
Regulation of cyclic AMP response-element binding-protein (CREB) by Gq/11-protein-coupled receptors in human SH-SY5Y neuroblastoma cells
DOI:10.1016/j.bcp.2007.10.015
URL
PMID:2593902
[本文引用: 1]
Human SH-SY5Y neuroblastoma cells have been used to investigate mechanisms involved in CREB phosphorylation after activation of two endogenously expressed Gq/11-protein-coupled receptors, the M3 muscarinic acetylcholine (mACh) and B2 bradykinin receptors. Stimulation with either methacholine or bradykinin resulted in maximal increases in CREB phosphorylation within 1min, with either a rapid subsequent decrease (bradykinin) to basal levels, or a sustained response (methacholine). Inhibitor studies were performed to assess the involvement of a number of potential kinases in signalling to CREB phosphorylation. Removal of extracellular Ca2+, inhibition of Ca2+/calmodulin-dependent protein kinase II and down-regulation of protein kinase C (PKC) resulted in reduced CREB phosphorylation after both M3 mACh and B2 bradykinin receptor activation. In contrast, inhibition of MEK1/2 by U0126 resulted in significantly reduced CREB phosphorylation levels after B2 bradykinin, but not M3 mACh receptor activation. In addition, we demonstrate that maintained phosphorylation of CREB is necessary for CRE-dependent gene transcription as the M3 mACh, but not the B2 bradykinin receptor activates both a recombinant CRE-dependent reporter gene, and the endogenous c-Fos gene. These data highlight the involvement of multiple, overlapping signalling pathways linking these endogenous Gq/11-coupled metabotropic receptors to CREB and emphasize the importance of the duration of signalling pathway activation in converting a CREB phosphorylation event into a significant change in transcriptional activity.
Protein interactome reveals converging molecular pathways among autism disorders
DOI:10.1126/scitranslmed.3002166
URL
PMID:21653829
[本文引用: 1]
To uncover shared pathogenic mechanisms among the highly heterogeneous autism spectrum disorders (ASDs), we developed a protein interaction network that identified hundreds of new interactions among proteins encoded by ASD-associated genes. We discovered unexpectedly high connectivity between SHANK and TSC1, previously implicated in syndromic autism, suggesting that common molecular pathways underlie autistic phenotypes in distinct syndromes. ASD patients were more likely to harbor copy number variations that encompass network genes than were control subjects. We also identified, in patients with idiopathic ASD, three de novo lesions (deletions in 16q23.3 and 15q22 and one duplication in Xq28) that involve three network genes (NECAB2, PKM2, and FLNA). The protein interaction network thus provides a framework for identifying causes of idiopathic autism and for understanding molecular pathways that underpin both syndromic and idiopathic ASDs.
Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci
DOI:10.1016/j.neuron.2015.09.016
URL
PMID:4624267
[本文引用: 1]
Through analysis of de novo mutations in autism spectrum disorder (ASD), Sanders et02al. find that small deletions, but not large deletions/duplications, contain one critical gene. Combining CNV and sequencing data, they identify 6 loci and 65 genes associated with ASD.
De novo mutations revealed by whole-exome sequencing are strongly associated with autism
Solving the autism puzzle a few pieces at a time
DOI:10.1016/j.neuron.2011.05.025
URL
PMID:21658575
[本文引用: 1]
In this issue, a pair of studies (Levy et02al. and Sanders et02al.) identify several de novo copy-number variants that together account for 5%–8% of cases of simplex autism spectrum disorders. These studies suggest that several hundreds of loci are likely to contribute to the complex genetic heterogeneity of this group02of disorders. An accompanying study in this issue (Gilman et02al.), presents network analysis implicating these CNVs in neural processes related to synapse development, axon targeting, and neuron motility.
Factor analysis of restricted and repetitive behaviors in autism using the autism diagnostic interview-r
DOI:10.1023/A:1025321707947
URL
PMID:14518620
The current study examined the factor structure of restricted and repetitive behaviors (RRB) in children with autism. Factor extraction procedures of 12 items from the Autism Diagnostic Interview-Revised ( ADI-R ) were applied in N = 207 individuals with autism. Two interpretable factors were identified: Factor 1— repetitive sensory motor actions and Factor 2— resistance to change . There was a significant negative correlation between an index of level of adaptive functioning and Factor 1. Intraclass correlations were not significant for either factor in a subset of families with two or more siblings with autism (multiplex). No differences in scores were apparent for either factor when multiplex families and families containing only one affected individual with autism (singleton) were compared. RRB in autism are represented by two distinct factors which may reflect two separate groups within autism. Defining subgroups within autism will allow for reduction of clinical heterogeneity and enhance our ability to dissect the genetic etiology of this complex disorder.
Subtelomeric deletions of 1q43q44 and severe brain impairment associated with delayed myelination
DOI:10.1038/jhg.2012.77
URL
PMID:22718018
[本文引用: 2]
Journal of Human Genetics, official journal of the Japan Society of Human Genetics, publishes original articles and reviews on all aspects of human genetics, including medical genetics and genomics
Familial duplication/deletion of 1q42.13q43 as meiotic consequence of an intrachromosomal insertion in chromosome 1
DOI:10.1159/000485226
URL
PMID:29258113
[本文引用: 1]
Abstract Rearrangements of the region 1q42.13q43 are rare, with only 7 cases reported to date. The imbalances described are usually the result of inherited translocations with other chromosomes. Moreover, few cases of both inter- and intrachromosomal deletions/duplications detected cytogenetically have been described. We report the molecular cytogenetic characterization of an inverted insertion involving the region 1q42.13q43 and segregating in 2 generations of a family. The deletion and the duplication of the same segment were detected in 2 affected family members. SNP array analysis showed the familial origin of the deletion/duplication due to the occurrence of a crossing-over during meiosis. Our report underlines the importance of determining the correct origin of chromosomal aberrations using different molecular cytogenetic tests in order to provide a good estimation of the reproductive risk for the members of the family.
CREB and memory
Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism
RYR2, PTDSS1 and AREG genes are implicated in a Lebanese population- based study of copy number variation in autism. Scientific Reports,
DOI:10.1038/srep19088
URL
PMID:26742492
[本文引用: 1]
Autism Spectrum Disorders (ASDs) are a group of neurodevelopmental disorders characterized by ritualistic-repetitive behaviors and impaired verbal and non-verbal communication. Objectives were to determine the contribution of genetic variation to ASDs in the Lebanese. Affymetrix Cytogenetics Whole-Genome 2.7 M and CytoScan鈩D Arrays were used to detect CNVs in 41 Lebanese autistic children and 35 non-autistic, developmentally delayed and intellectually disabled patients. 33 normal participants were used as controls. 16de novoCNVs and 57 inherited CNVs, including recognized pathogenic 16p11.2 duplications and 2p16.3 deletions were identified. A duplication at 1q43 classified as likely pathogenic encompassesRYR2as a potential ASD candidate gene. This previously identified CNV has been classified as both pathogenic, and, of uncertain significance. A duplication of unknown significance at 10q11.22, proposed as a modulator for phenotypic disease expression in Rett syndrome, was also identified. The novel potential autism susceptibility genesPTDSS1 and AREGwere uncovered and warrant further genetic and functional analyses. Previously described and novel genetic targets in ASD were identified in Lebanese families with autism. These findings may lead to improved diagnosis of ASDs and informed genetic counseling, and may also lead to untapped therapeutic targets applicable to Lebanese and non-Lebanese patients.
Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes
DOI:10.1136/jmedgenet-2014-102803
URL
PMID:25527629
[本文引用: 1]
Germline mutations in the phosphatase PTEN are associated with diverse human pathologies, including tumour susceptibility, developmental abnormalities and autism, but any genotype-phenotype relationships are poorly understood.We have studied the functional consequences of seven PTEN mutations identified in patients diagnosed with autism and macrocephaly and five mutations from severe tumour bearing sufferers of PTEN hamartoma tumour syndrome (PHTS).All seven autism-associated PTEN mutants investigated retained the ability to suppress cellular AKT signalling, although five were highly unstable. Observed effects on AKT also correlated with the ability to suppress soma size and the length and density of dendritic spines in primary neurons. Conversely, all five PTEN mutations from severe cases of PHTS appeared to directly and strongly disrupt the ability to inhibit AKT signalling.Our work implies that alleles causing incomplete loss of PTEN function are more commonly linked to autism than to severe PHTS cases.
The emerging biology of autism spectrum disorders
Genetic influences on the broad spectrum of autism: Study of proband-ascertained twins. American Journal of Medical
DOI:10.1002/ajmg.b.30740
URL
PMID:18361421
[本文引用: 1]
An investigation of genetic structures underlying autistic traits was performed with samples from twins for which at least one proband had been ascertained as having autism spectrum disorders (ASDs) in our catchment area. In order to adjust for recent concepts of autism, we employed criteria for the broad spectrum of disease and the childhood autism rating scale (CARS) for quantitative assessment. The CARS test was performed on 45 twin pairs (19 monozygotic, 26 dizygotic) detected with a regional routine screening system. The obtained CARS scores were subjected to structural equation modeling (SEM), incorporating sex differences for each causal influence ascertainment correction, using the Mx software. A best fitting model of causal influences on autistic traits measured continuously, incorporating additive genetic (A) and non-shared environmental influences (E), was generated. With this AE model, the estimated heritability was 0.73 for males and 0.87 for females, based on the continuous CARS scores. There was no evidence for the existence of sex-specific genetic influences. Autistic traits were highly heritable in twins with even broad spectrum of autism, corresponding to the results of early studies based on classical autism. Additive genetic factors were more influential in females than males. 2008 Wiley-Liss, Inc.
Accumulation of c-fos mRNA in rat hippocampus during acquisition of a brightness discrimination
DOI:10.1016/0163-1047(90)91366-J
URL
PMID:2122879
[本文引用: 1]
Training rats to attain a foot-shock-motivated brightness discrimination in a Y-maze results in an early and transient increase of hippocampal c-fos mRNA levels. Maximal accumulation was observed immediately after training, returning to basal levels during the following 2 h. A similar increase was obtained when rats were subjected to a pseudotraining with an equal number of runs, but with random pairing of the choice of bright and dark alleys with foot shock. It is suggested that induction of hippocampal c-fos mRNA expression is a necessary, but not sufficient, prerequisite for the formation of long-term memory trace. This early gene expression seems rather to correspond to an initial stage induced by complex stimulus presentation of both the training and the pseudotraining procedure. The subsequent late synthesis or processing of target proteins finally contributing to the formation of a permanent trace requires the action of further convergent signals to principal cells, probably mediating reward or emotional influences.
Loss of [3h]4-damp binding to muscarinic receptors in the orbitofrontal cortex of alzheimer's disease patients with psychosis
DOI:10.1007/s00213-008-1124-9
URL
PMID:18373228
[本文引用: 1]
Neuropsychiatric behaviours in Alzheimer's disease (AD) patients have been associated with neocortical alterations of presynaptic cholinergic and muscarinic M2 receptor markers. In contrast, it is unclear whether non-M2 muscarinic receptors have a role to play in AD behavioural symptoms.To correlate the alterations of neocortical postsynaptic muscarinic receptors with clinical features of AD.[(3)H]4-DAMP were used in binding assays with lysates of Chinese hamster ovary (CHO) cells stably transfected with M1-M5 receptors. [(3)H]4-DAMP was further used to measure muscarinic receptors in the postmortem orbitofrontal cortex of aged controls and AD patients longitudinally assessed for cognitive decline and behavioural symptoms.[(3)H]4-DAMP binds to human postmortem brain homogenates and M1-, M3-, M4- and M5-transfected CHO lysates with subnanomolar affinity. Compared to the controls, the [(3)H]4-DAMP binding density is reduced only in AD patients with significant psychotic symptoms. The association between reduced [(3)H]4-DAMP binding and psychosis is independent of the effects of dementia severity or neurofibrillary tangle burden.This study suggests that the loss of non-M2 muscarinic receptors in the orbitofrontal cortex may be a neurochemical substrate of psychosis in AD and provides a rationale for further development of muscarinic receptor ligands in AD pharmacotherapy.
Autism genetics: Opportunities and challenges for clinical translation
DOI:10.1038/nrg.2017.4
URL
PMID:28260791
[本文引用: 1]
Abstract Genetic studies have revealed the involvement of hundreds of gene variants in autism. Their risk effects are highly variable, and they are frequently related to other conditions besides autism. However, many different variants converge on common biological pathways. These findings indicate that aetiological heterogeneity, variable penetrance and genetic pleiotropy are pervasive characteristics of autism genetics. Although this advancing insight should improve clinical care, at present there is a substantial discrepancy between research knowledge and its clinical application. In this Review, we discuss the current challenges and opportunities for the translation of autism genetics knowledge into clinical practice.
Intrastriatal injection of a muscarinic receptor agonist and antagonist regulates striatal neuropeptide mRNA expression in normal and amphetamine-treated rats
DOI:10.1016/S0006-8993(96)01244-9
URL
PMID:9067445
[本文引用: 1]
Systemic administration of the muscarinic receptor antagonist, scopolamine, augments, whereas the muscarinic receptor agonist, oxotremorine, attenuates behaviors (locomotion and stereotypies) and preprodynorphin (PPD) and substance P (SP) gene expression in striatonigral neurons induced by the indirect dopamine receptor agonist, amphetamine (AMPH). In contrast, systemic scopolamine blocks, whereas oxotremorine augments, AMPH-stimulated preproenkephalin (PPE) gene expression in striatopallidal neurons. This study investigated the site of action of these effects by administering scopolamine and oxotremorine directly into the striatum and assessing the expression of neuropeptide mRNAs with quantitative in situ hybridization. Unilateral injection of scopolamine into the dorsal striatum augmented, and oxotremorine attenuated, AMPH (2.5 mg/kg, i.p.)-stimulated behaviors. Intrastriatal scopolamine at a concentration of 62 mM, but not 6.2 mM, increased basal levels of PPD and SP mRNAs in the dorsal striatum. In addition, both 6.2 and 62 mM scopolamine significantly augmented AMPH-stimulated PPD and SP mRNA levels. Intrastriatal infusion of 1.6 or 8.1 mM oxotremorine did not alter basal levels of striatal PPD and SP mRNAs. However, both concentrations of oxotremorine completely blocked AMPH-stimulated SP mRNA and oxotremorine at 8.1 mM blocked AMPH-stimulated PPD mRNA. In contrast, PPE induction by AMPH was blocked by 62, but not 6.2, mM scopolamine. Both concentrations of oxotremorine tended to augment basal and AMPH-stimulated PPE mRNA in the dorsal striatum but the trend was not significant. These data demonstrate an inhibition of striatonigral, and facilitation of striatopallidal, gene expression through activation of local striatal muscarinic receptors, which is consistent with the changes seen after systemic administration of muscarinic agents. Therefore, muscarinic cholinergic regulation of basal and stimulated expression of neuropeptide mRNA is processed within the striatum.
Alzheimer's disease and senile dementia: Loss of neurons in the basal forebrain
DOI:10.1126/science.7058341
URL
PMID:7058341
[本文引用: 1]
Recent evidence indicates that the nucleus basalis of Meynert, a distinct population of basal forebrain neurons, is a major source of cholinergic innervation of the cerebral cortex. Postmortem studies have previously demonstrated profound reduction in the presynaptic markers for cholinergic neurons in the cortex of patients with Alzheimer's disease and senile dementia of the Alzheimer's type. The results of this study show that neurons of the nucleus basalis of Meynert undergo a profound (> 75 percent) and selective degeneration in these patients and provide a pathological substrate of the cholinergic deficiency in their brains. Demonstration of selective degeneration of such neurons represents the first documentation of a loss of a transmitter-specific neuronal population in a major disorder of higher cortical function and, as such, points to a critical subcortical lesion in Alzheimer's patients.
Language, social, and cognitive impairments in autism and severe mental retardation
DOI:10.1007/BF01531339
URL
PMID:6927697
[本文引用: 1]
An epidemiological study is outlined that shows that Kanner's syndrome is one group among a wider range of children, all with impairment of social interaction, communication, and imagination. Most, but not all, children with this triad of impairments are severely mentally retarded, although severe retardation also occurs in those who are sociable and communicative. It is hypothesized that the socially impaired lack certain abilities that are inborn in normal children and the sociable mentally retarded: namely, the capacity to produce and monitor the normal speciesspecific preverbal sounds, the drive to explore the environment and form concepts to explain experiences, and the ability to recognize that other human beings are of special interest and importance. A possible neurological basis for these problems is briefly considered .
Evidence for ASD recurrence rates and reproductive stoppage from large UK ASD research family databases
DOI:10.1002/aur.1414
URL
PMID:25273900
[本文引用: 1]
Abstract Following a diagnosis of a developmental disorder such as autism spectrum disorder (ASD) in early childhood, parents may decide to have fewer children than previously planned. The tendency for families to halt reproduction after receiving a diagnosis for one child is known as reproductive stoppage. Stoppage may lead to an underestimate of recurrence risk estimates of parents having more than one child with ASD. Using two large UK ASD family databases, we investigated recurrence rates for ASD and evidence for reproductive stoppage for both ASD and undiagnosed ASD/broader autism phenotype in a subgroup of families. Reproductive stoppage was tested for using the Mann-Whitney U-test to disprove the null hypothesis that affected and nonaffected children were distributed randomly by birth order. Dahlberg's later-sib method was used to estimate recurrence risk and take stoppage into account. Data were available from 299 families (660 children) including 327 with ASD. Ten percent of the complete families had more than one child with an ASD. Using Dahlberg's later-sib method, the recurrence risk for ASD was 24.7% overall and 50.0% in families with two or more older siblings with ASD. Children with ASD were born significantly later in families than those without ASD in all sibship combinations. This study shows strong evidence that ASD is associated with reproductive stoppage. These data have important implications for family planning and genetic counseling. 2014 International Society for Autism Research, Wiley Periodicals, Inc.
Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean
DOI:10.1038/35065604
URL
PMID:11242080
[本文引用: 2]
Abstract Members of the muscarinic acetylcholine receptor family (M1-M5) have central roles in the regulation of many fundamental physiological functions. Identifying the specific receptor subtype(s) that mediate the diverse muscarinic actions of acetylcholine is of considerable therapeutic interest, but has proved difficult primarily because of a lack of subtype-selective ligands. Here we show that mice deficient in the M3 muscarinic receptor (M3R-/- mice) display a significant decrease in food intake, reduced body weight and peripheral fat deposits, and very low levels of serum leptin and insulin. Paradoxically, hypothalamic messenger RNA levels of melanin-concentrating hormone (MCH), which are normally upregulated in fasted animals leading to an increase in food intake, are significantly reduced in M3R-/- mice. Intra-cerebroventricular injection studies show that an agouti-related peptide analogue lacked orexigenic (appetite-stimulating) activity in M3R-/- mice. However, M3R-/- mice remained responsive to the orexigenic effects of MCH. Our data indicate that there may be a cholinergic pathway that involves M3-receptor-mediated facilitation of food intake at a site downstream of the hypothalamic leptin/melanocortin system and upstream of the MCH system.
Social approach behaviors are similar on conventional versus reverse lighting cycles, and in replications across cohorts, in BTBR T+ tf/J, C57BL/6J, and vasopressin receptor 1B mutant mice
DOI:10.3389/neuro.08/001.2007
URL
PMID:2525856
[本文引用: 1]
Mice are a nocturnal species, whose social behaviors occur primarily during the dark phase of the circadian cycle. However, laboratory rodents are frequently tested during their light phase, for practical reasons. We investigated the question of whether light phase testing presents a methodological pitfall for investigating mouse social approach behaviors. Three lines of mice were systematically compared. One cohort of each line was raised in a conventional lighting schedule and tested during the light phase, under white light illumination; another cohort was raised in a reverse lighting schedule and tested during their dark phase, under dim red light. Male C57BL/6J (B6) displayed high levels of sociability in our three-chambered automated social approach task when tested in either phase. BTBR T+ tf/J (BTBR) displayed low levels of sociability in either phase. Five cohorts of vasopressin receptor subtype 1b (Avpr1b) null mutants, heterozygotes, and wildtype littermate controls were tested in the same social approach paradigm: three in the dark phase and two in the light phase. All three genotypes displayed normal sociability in four out of the five replications. In the juvenile play test, testing phase had no effect on play soliciting behaviors in Avpr1b mice, but had modest effects on nose sniff and huddling. Taken together, these findings indicate that testing phase is not a crucial factor for studying some forms of social approach in juvenile and adult mice.
Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus
DOI:10.1002/jnr.22546
URL
PMID:21162125
[本文引用: 1]
Fragile X syndrome (FXS) is a monogenic mental retardation syndrome that frequently includes autism. The Fmr1-knockout (Fmr1-KO) mouse, like FXS-affected individuals, lacks the fragile X mental retardation protein (FMRP) and models autism as well as FXS. Limited human data and several mouse models have implicated the hippocampal dentate gyrus (DG) in autism. We therefore investigated whether the Fmr1-KO mouse exhibited functional changes in DG. We found diminished medial perforant path-granule cell long-term potentiation (LTP), complementing previous investigations of synaptic plasticity in Fmr1-KO demonstrating impaired LTP in CA1, neocortex, and amygdala and exaggerated long-term depression in CA1. We also found that peak amplitude of NMDA receptor-mediated excitatory postsynaptic currents (EPSCs) was smaller in Fmr1-KO than control. AMPA receptor-mediated EPSCs were comparable in the two strains, yielding a lower NMDA/AMPA ratio in Fmr1-KO mice and suggesting one mechanism by which absent FMRP might contribute to diminished LTP. The clinical hallmarks of autism include both excessive adherence to patterns and impaired detection of socially important patterns. The DG has a putative role in pattern separation (for time, space, and features) that has been attributed to granule cell number, firing rates, adult neurogenesis, and even perforant path LTP. DG also contributes to pattern completion in CA3 via its mossy fiber efferents, whose terminals include abundant FMRP in ragile X granules. Together with the present data, these observations suggest that DG is a candidate region for further investigation in autism and that the Fmr1-KO model may be particularly apt. 2010 Wiley-Liss, Inc.
Enhanced endocannabinoid signaling elevates neuronal excitability in fragile X syndrome
Loss of MeCP2 in cholinergic neurons causes part of RTT-like phenotypes via alpha7 receptor in hippocampus
DOI:10.1038/cr.2016.48
URL
PMID:27103432
[本文引用: 1]
Author(s): Zhang, Y; Cao, SX; Sun, P; He, HY; Yang, CH; Chen, XJ; Shen, CJ; Wang, XD; Chen, Z; Berg, DK; Duan, S; Li, XM | Abstract: 2016 IBCB, SIBS, CAS All rights reserved. Mutations in the X-linked MECP2 gene cause Rett syndrome (RTT), an autism spectrum disorder characterized by impaired social interactions, motor abnormalities, cognitive defects and a high risk of epilepsy. Here, we showed that conditional deletion of Mecp2 in cholinergic neurons caused part of RTT-like phenotypes, which could be rescued by re-expressing Mecp2 in the basal forebrain (BF) cholinergic neurons rather than in the caudate putamen of conditional knockout (Chat-Mecp2-/y) mice. We found that choline acetyltransferase expression was decreased in the BF and that 7 nicotine acetylcholine receptor signaling was strongly impaired in the hippocampus of Chat-Mecp2-/y mice, which is sufficient to produce neuronal hyperexcitation and increase seizure susceptibility. Application of PNU282987 or nicotine in the hippocampus rescued these phenotypes in Chat-Mecp2-/y mice. Taken together, our findings suggest that MeCP2 is critical for normal function of cholinergic neurons and dysfunction of cholinergic neurons can contribute to numerous neuropsychiatric phenotypes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}